

N-Acetilcisteína: relación entre mecanismos de acción y probables usos en trastornos neuropsiquiátricos
Resumen
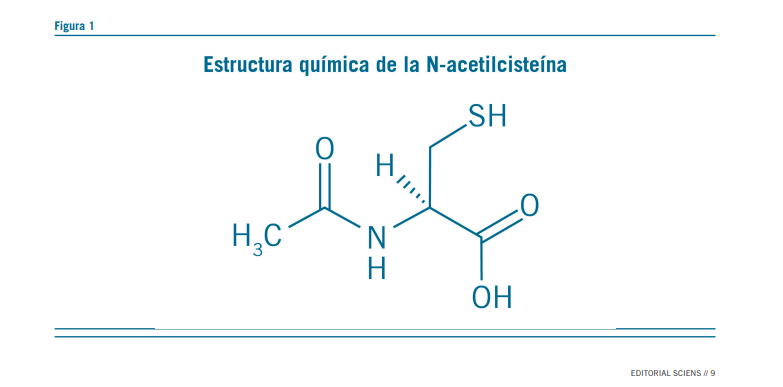
La N-Acetilcisteína es un derivado del aminoácido no esencial cisteína, en la que el grupo acetilo está unido al átomo de nitró- geno. Es un precursor del antioxidante glutatión, a su vez modulador de las vías glutamatérgicas neurotróficas e inflamatorias.
En este artículo se relaciona el mecanismo de acción de esta molécula con su probable utilidad en trastornos neurológicos y psiquiátricos, a saber: síndrome de Down, enfermedades espinocerebelosas, epilepsia mioclónica de Unverricht–Lundbor, adrenoleucodistrofia ligada al X, disquinesia tardía, enfermedades de Parkinson, Huntinghton y Alzheimer, hemorragia subarac- noidea, ELA, isquemia cerebral focal y lesión cerebral traumática, prevención de daños cerebrales fetales tras infección / infla- mación materna, acidemia glutárica tipo I, encefalopatía por fallo renal y diabética, esquizofrenia, trastorno bipolar, adicciones, tricotilomanía, onicofagia, trastorno de excoriación, juego patológico y auto injuria no suicida.
Para dicho fin se realizaron búsquedas en distintas bases de datos: Pub-Med, Google Académico, Medes, Lilacs, DOAJ, Psy- cINFO y Psiquiatría.com, con las palabras clave descriptas y sus combinaciones.
Palabras clave
N-acetilcisteína – Psiquiatría – Neurología – Mecanismo de acción – Neuroinflamación.
Angemi JA. “N-Acetilcisteína: relación entre mecanismos de acción y probables usos en trastornos neuropsiquiátricos”. Psicofarmacología 2022;8:15. Puede consultar otros artículos publicados por los autores en la revista Psicofarmacología en sciens.com.ar
La N-Acetilcisteína (NAC) es un derivado del aminoácido no esencial cisteína, en la que el grupo acetilo está unido al áto- mo de nitrógeno (Figura 1). Es un precursor del antioxidante glutatión (GSH), a su vez modulador de las vías glutamatérgi- cas neurotróficas e inflamatorias (1, 2).
Originalmente, su uso se basaba en sus propiedades mu- colíticas y como antídoto de la intoxicación por paracetamol, pero en las últimas décadas se conoce su utilidad en varias
entidades psiquiátricas y neurológicas. Activa los sistemas antioxidantes y de óxido nítrico durante el estrés, las infec- ciones, los ataques tóxicos y las condiciones inflamatorias (3). Tiene escasos efectos adversos, no vinculado a dosis, como náuseas, vómitos, diarrea, constipación y gastralgia, contrain- dicándose en úlcera péptica sangrante. Este perfil resulta muy
favorable para su uso (4).
Farmacocinética
Tras la administración de NAC por vía oral la absorción se produce rápidamente, alcanzando concentraciones séricas máximas en 2 a 3 horas. Luego de una dosis de 200 a 400 mg el pico plasmático es de 0,35 a 4 mg/L a los 60-120 minutos. Se desacetila y circula en forma libre ligada a las proteínas plasmáticas. La biodisponibilidad es de un 5 %, de- bido a la n-desacetilación en mucosa intestinal y primer paso hepático. Su vida media es de aproximadamente 6 h (11 en niños). Ésta aumenta un 80 % en insuficiencia renal grave y en cirrosis. También se observa un incremento en los niveles plasmáticos de cisteína y glutatión, aspecto relacionado con su propio mecanismo de acción. Difunde de forma rápida a los líquidos extracelulares, localizándose principalmente a nivel de la secreción bronquial. Su volumen de distribución es de 0,33 a 0,47 l/Kg. Se degrada principalmente en hígado (70
%), riñones y pulmones Su eliminación es renal en un 30 %
(clearence de 0,19 a 0,211 L/h) y sus principales metabolitos los aminoácidos cistina y cisteína. Un 50 % circula unida a proteínas por uniones disulfuro (lábiles) o enlaces peptídicos covalentes luego de 4 horas de su administración. Su princi- pal producto de excreción es el sulfato inorgánico (2, 5).
La NAC es permeable a la membrana celular, por lo que no requiere ser transportada como la cisteína por el sistema ala- nina-serina-cisteína (ASC), un sistema de transporte de ami- noácidos neutros dependiente de Na. Dentro de la célula es rápidamente hidrolizada y convertida en cisteína (6).
Luego de 2 horas de la administración oral de NAC a ratas, la concentración más alta se observó en el riñón y el hígado, seguido de las glándulas suprarrenales, los pulmones, el bazo, la sangre, los músculos y el cerebro. La NACA (NAC acetila- da), es un metabolito activo que atraviesa barrera hematoen- cefálica (2, 7).
Mecanismos de acción
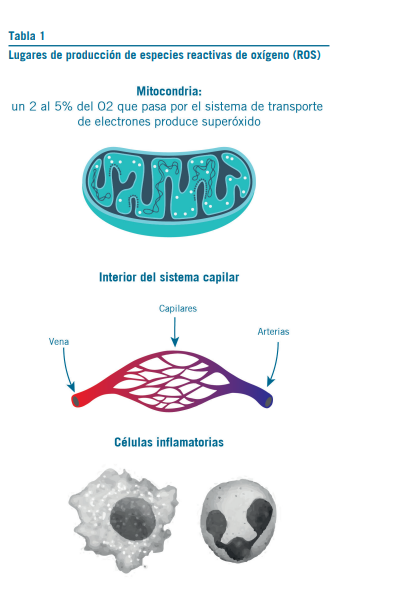
NAC es un tiol precursor de L-cisteína y GSH reducido. Es fuente de grupos sulfhidrilo en las células y un potente eli- minador de radicales libres, ya que interactúa con especies reactivas de oxígeno (ROS) como OH y H2O2 (ver lugares de producción en Tabla 1). Estos provocan daños en ADN, lípidos y proteínas por oxidación y peroxidación (1, 2).
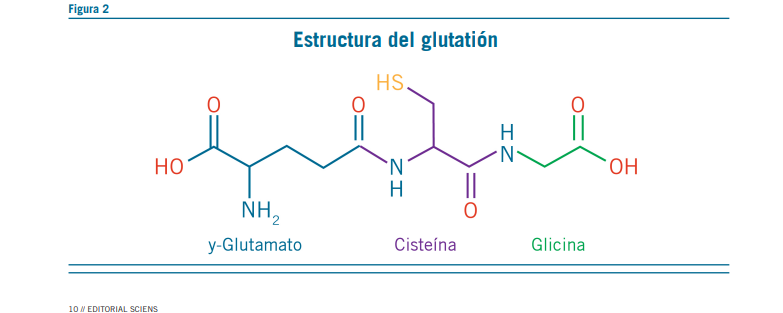
El GSH está compuesto por glutamato, glicina y cisteína (Figura 2), siendo la disponibilidad de esta última la que li- mita su síntesis en situaciones de estrés, por ser la que se encuentra en bajas concentraciones intracelulares. De esta forma, el NAC previene la depleción del GSH. Este posee va- rias acciones sobre el proceso de óxido-reducción (Tabla 2) (2, 8). En su síntesis intervienen las enzimas c-glutamincis- teína sintetasa y la GSH sintetasa. La primera es inhibida a través de un mecanismo de feedback por el mismo GSH (9).
NAC redujo la activación (fosforilación) temprana de MLK (Mitogen-activated protein kinase) 3 y 7, y JNK3 (c-Jun N-ter- minal Kinase) inducida por el beta-amiloide (Aß) y proporcio- nó una capacidad neuroprotectora débil en la apoptosis pro- ducto de dicha reacción (10). En ratas se demostró que inhibe a MLK-3 en las primeras etapas de isquemia hipocámpica, mejorando la reperfusión (11).
Inhibe a la VLDL (prooxidante) y a la urotensina (vasocons- trictor), evitando así la proliferación de músculo liso vascu- lar (VSMC), previniendo la ateroesclerosis. También inhibe el PDGF (Factor de crecimiento derivado de plaquetas) sérico y la activación de la cinasa regulada simple extracelular esti- mulada por trombina (ERK2), la JNK1 y la activación de la proteína cinasa activada por mitógeno p38 (MAPK), así como la expresión de c-Fos (70 %), genes c-Jun (50 %) y JunB (70 %), lo que sugiere mecanismos redox para los efectos protectores de la NAC en pacientes con factores de riesgo vascular importantes. Provoca down regulation del receptor de
angiotensina II tipo 1, antagonizando el efecto del ARNm de angiotensina II mediada por ROS, contribuyendo a la reduc- ción de la tensión arterial (12, 13).
Bloquea la producción de superóxido estimulada por sero- tonina y la fosforilación de ERK-MAPK en VSMC, reduciendo la proliferación de la capa íntima, e inhibe la inducción por benzopireno (producto de la combustión del cigarrillo) de la ciclooxigenasa 2 (14, 15).
Inhibe en un 60 % la expresión potenciada por homocisteí- na del receptor lox-1 de ox-LDL en el endotelio y la actividad y expresión de MMP-9 (gelatinasa B) en células espumosas derivadas de macrófagos cargados de lípidos, lo que demues- tra su potencial antioxidante sobre placas ateromatosas. Un ensayo clínico demostró que la administración oral diaria de NAC a una dosis de 1,2 g aumentó el GSH y disminuyó los niveles de la molécula de adhesión de células vasculares plas- máticas 1 (VCAM-1) en pacientes diabéticos no insulinode- pendientes (16, 17, 18).
Sus propiedades antiapoptóticas se demuestran por varios mecanismos. Aumenta la concanavalina-A inducida por mito- sis, impidiendo la apoptosis de linfocios B. Bloquea la apop- tosis de células endoteliales inducida por lipopolisacáridos (LPS). Bloquea la producción de H2O2 inducida por Ox-LDL
y la apoptosis de las células endoteliales de la vena umbilical
humana. Inactiva la producción de VSMC a través de la inhibi- ción de la producción de colágeno inducida por homocisteína. Previene la muerte celular inducida por FNT y trombina. In vitro inhibe la apoptosis inducida por arabinósidos a través de la re- ducción de ROS. También disminuye la producción de GFAP (proteína ácida fibrilar glial) tanto en corteza como en cuerpo estriado y la Iba-1 (proteína de unión al Ca en microglía y macrófagos que induce fagocitosis) en este último (2,19, 20, 21, 22, 23).
Ejerce un efecto modulador sobre NF-kB, aumentando la acción del I-Kb (inhibidor de NF-Kb) impidiendo su trasloca- ción al núcleo celular. Teniendo en cuenta que el NF-Kb tiene un papel cardinal en la regulación y expresión de genes de respuesta al estrés bajo desafíos inflamatorios y oxidativos, el
resultado final es la inhibición de la producción de citoquinas proinflamatorias (7).
Aumenta la concanavalina-A inducida por mitogénesis al tiempo que inhibe la apoptosis por linfocitos B y la apoptosis de células endoteliales mediada por LPS (19, 24).
Disminuye la acumulación de Zn (tiene sitios de unión para metales traza que incluyen también a Cu y Mg). y la sobreac- tivación del TRPM2 (receptor potencial transitorio de melas- tatina 2). El mismo es un canal catiónico no selectivo, cuya actividad es activada por ROS, TNF-alfa y H2O2. Se encuen- tra distribuido en todo el SNC, predominando en hipocampo, estriado y cortex. Permite el paso de cationes divalentes como Ca y Mg (25, 26).
La pérdida de estructura como el alfa-hélice de algunas pro- teínas (inducido por toxicidad) provocan acumulación excesi- va de las mismas, produciendo enfermedades neurodegene- rativas como el Parkinson (EP), Alzheimer (EA) y enfermedad de Huntington (EH). Se ven más expuestas las proteínas que presentan elevadas repeticiones de aminoácidos en su estruc- tura, como por ejemplo la poliglutamina en la EH. Las protei- nopatías relacionadas con sustancias tóxicas con pérdida de GSH podrían tener una buena respuesta a la NAC al revertir esta pérdida de GSH y prevenir esta toxicidad. Este mecanis- mo estaría regulado por chaperonas (proteínas responsables del plegamiento de proteínas) (27).
Utilidad en enfermedades neuropsiquiátricas
Teniendo en cuenta que el estrés oxidativo y el rol del GSH juegan un papel fundamental en las patologías degenerativas neurológicas y psiquiátricas, cada vez hay más estudios que plantean la utilidad del NAC como tratamiento de estas.
A continuación, se hace una mención de dichas entidades junto a los mecanismos fisiopatológicos involucrados como posibles targets de NAC.
Enfermedades espinocerebelosas
Cursan con elevado estrés oxidativo. Se vieron mejoras en
disartria, ataxia, movimientos oculomotores, propiocepción y sensibilidad al dolor con uso de NAC (2, 28).
Epilepsia mioclónica de Unverricht–Lundbor
Es un trastorno autosómico recesivo que se desarrolla en- tre los 6 y los 15 años con mioclonía sensible a estímulos y convulsiones tónico-clónicas seguidas de síndrome cerebelo- so progresivo (2, 28). Teniendo en cuenta que en las crisis epilépticas existe elevado estrés oxidativo y disfunción mito- condrial, Devi y colaboradores (29) comunican la efectividad de la combinación de divalproato de Na y NAC en ratas a las cuales se indujeron convulsiones por electroshock.
Síndrome de Down
Hay un 50 % de sobreexpresión del gen de la superóxido dimutasa (SOD) y mayor prevalencia de EA en edades tem- pranas (30).
Adrenoleucodistrofia ligada al X (X-ALD)
Es un trastorno peroxisomal que produce desmielinización cerebral y disfunción axonal de la médula espinal por estrés oxidativo. Conduce a una paraplejía espástica, insuficiencia suprarrenal y ocasionalmente insuficiencia testicular. Se observa clínicamente déficit cognitivo moderado seguido de agudeza visual disminuida, sordera central, ataxia cerebelo- sa, hemiplejía, convulsiones y demencia que conducen a un estado neurovegetativo o la muerte al cabo de varios años. Su herencia es de penetrancia completa en varones y del 60
% de las mujeres heterocigotas Padilha Marchetti et al. (31) comunicaron efectividad de la NAC en estudios in vitro por incremento de GSH (disminuidos en la enfermedad) y dismi- nución del estrés oxidativo. Aumenta la expresión de antioxi- dantes como la enzima hemo oxigenasa y su efectora ferritina (unas 4 y 160 veces respectivamente) (32, 33).
Disquinesia tardía
Al bloquear los receptores de dopamina (DA), los neuro- lépticos pueden provocar la acumulación de dicho neuro- transmisor en los ganglios basales, lo que luego aumenta la producción de radicales libres. Los ganglios basales son extre- madamente sensibles a la sobrecarga de éstos (34).
EH
El principal evento involucrado es la disfunción mitocondrial. La NAC disminuiría la lesión en cuerpo estriado (35). Sandhir et al. (36) revirtieron con NAC el daño mitocondrial producido por ácido nitropropiónico en un modelo de EH en ratas.
EP
Hay incremento de peroxidación y disminución de GSH. Dicha disminución es correlativa con la gravedad de la enfermedad. El estrés oxidativo aumenta la acumulación de alfa- sinucleína, con aumento de la formación de nitrotirosina y activación microglial. En sustancia negra hay elevación de Fe y neuromelanina, lo que incrementa el ROS. La acción de la MAO sobre las catecolaminas aumenta el H2O2. También se relaciona el uso de NAC con elevaciones del DAT-1 en caudado y putamen luego de 3 meses de administración medida con DAT-Scan (28, 37, 38).
EA
Hay aumento de peroxidación lipídica en corteza temporal y disminución de GSH hipocampal (2). El deterioro en dos transportadores de flujo de efecto de la barrera hematoence- fálica (BHE), la glicoproteína p (Pgp) y la proteína 1 relaciona- da con el receptor de lipoproteínas de baja densidad (LRP-1) contribuirían a la progresión de la EA por acumulación de Ab. Esto se desencadenaría por estrés oxidativo. NAC demostró protección contra la disfunción del transporte de Ab inducida por LPS en la BHE a través de un mecanismo dependiente de LRP-1 e independiente de Pgp (39).
Esclerosis múltiple (EM)
Hay aumento de expresión de TNF, con incremento de pro-
ducción de ROS. En modelos animales NAC inhibió el desa- rrollo de EM-like (2, 40).
Hemorragia subaracnoidea
Por vasoespasmo, se generan radicales libres que promue- ven la peroxidación lipídica y daño endotelial (2). Esclerosis lateral amiotrófica (ELA)
Se describieron mutaciones del gen de la superóxido-dimu- tasa (SOD1) en casos de transmisión familiar. En casos de presentación esporádica no se encontraron alteraciones de la SOD1, pero sí una marcada disminución de la actividad de la GSH-reductasa (2, 41).Isquemia cerebral focal y lesión cerebral traumática
Hay alteración mitocondrial, con aumento de formación de ROS. Aumento de mediadores humorales relacionados a isquemia: opioides, adenosina, ON, bradiquininas, catecolaminas, proteínas de shock térmico, hemooxigenasa, TNF-alfa, angiotensina y prostaglandinas. Liberación masiva de gluta- mato (neurotransmisor excitatorio), estimulando masivamente a los receptores NMDA. Esta acción presenta un efecto inhibidor del TNF-alfa, disminuye la secreción de EF-1(endoteli- na 1) por up regulaton del ARNm del pre-pro ET-1. También inhibe la regulación positiva de ET-1 mediante la inhibición de la activación transcripcional de la expresión del gen ET-1 mediada por NF-kappaB. Disminuye la formación de ATP y aumento de glucólisis anaeróbica (42, 43, 44). También hay liberación masiva de Zn en el espacio sináptico, con su posterior ingreso neuronal a través de transportadores catiónicos. Este metal, si bien se encuentra ampliamente distribuido en SNC, produce neurodegeneración hipocampal luego de daño isquémico, hipoglucémico y traumático. La isquemia a su vez produce activación del TRPM2 (Transient receptor potential cation cannel) que se encuentra altamente distribuido en el sistema nervioso central (neuronas y células gliales) y tiene una alta sensibilidad al daño oxidativo, contribuyendo a la activación de cascadas inflamatorias y neurodegenerativas. El NAC disminuye la acumulación de Zn y la sobreactivación del TRPM2 (25).
Prevención de daños cerebrales fetales tras infección/ infla- mación materna
La utilización de NAC profiláctica, administrada antes y después del LPS materno, redujo la lesión cerebral de las crías en ratas. Las crías de las madres tratadas con LPS ex- hibieron niveles de difusividad media, axial y radial signifi- cativamente mayores en la sustancia blanca y gris (medida en RNM con tensor de difusión), lo que concuerda con una lesión cerebral. Por el contrario, las crías de madres tratadas con LPS más NAC demostraron niveles de difudividad medios, axiales y radiales reducidos en la mayoría de las regiones. El tratamiento con NAC en la madre después de la inflamación materna influyó significativamente en la integridad de la mi- croestructura cerebral (45).
Acidemia glutárica tipo I (AG-1)
Es una enfermedad metabólica de herencia autosómica recesiva debido al déficit de la enzima mitocondrial gluta- ril-CoA-deshidrogenasa (GCDH) que interviene en el metabolismo de degradación de los aminoácidos lisina, hidroxilisina y triptófano. Esto provoca acumulación de ácido glutámico. En general debuta como una encefalopatía a los 3 a 6 meses de edad, conduciendo a una degeneración progresiva cortical y nigroestriatal. Provoca alteraciones motrices y cognitivas, en especial en memoria de procedimiento. Si bien el tratamiento de elección es la carnitina, la NAC demostró su utilidad al disminuir la activación de células gliales en respuesta a la acumulación de ácido glutámico, inhibiendo la peroxidación lipídica mediante la disminución del NGF, Bcl-2 (que inter- viene en el proceso de permeabilidad mitocondrial) y p75- NTR (receptor de neurotrofina). Todos los factores menciona- dos son proapoptóticos (23, 46).
Tabla 2
Acciones del glutatión
| Antioxidante |
| Desintoxicación de xenobióticos electrofílicos |
| Modulación de redox (reacción de oxidación-reducción) |
| Almacenamiento y transporte de cisteína |
| Regulación de la proliferación celular |
| Regulación de síntesis de ADN |
| Regulación de las respuestas inmunitarias |
| Regulación del metabolismo de leucotrienos y prostaglandinas |
Encefalopatía por fallo renal
La creatin kinasa (CK) estaría involucrada en la patogénesis de la encefalopatía urémica. Esa enzima contribuye a la ho- meostasis de la energía cerebral y es inhibida por los radicales libres que se producen por intixicación ureica. La actividad de CK fue inhibida en la corteza prefrontal y el hipocampo de ratas 12 h después de la isquemia renal. El tratamiento con NAC impidió tal efecto (47).
Encefalopatía diabética
Se caracteriza por déficits cognitivos, provocados por estrés oxidativo inducido por hiperglucemia a nivel mitocondrial. Disminuye la actividad de las enzimas NADH deshidrogenasa, succinato deshidrogenasa y citocromo oxidasa. En citoplasma hay aumento de citocromo c y caspasa 3 activada. Esto se tra- duce en inflamación mitocondrial y condensación de cromati- na en neuronas de ratas diabéticas. También hay aumento de compuestos carbonílicos reactivos como el metilglioxal, que lleva a una hiperproducción de ROS, con daño mitocondrial y alteraciones del endotelio microvascular (48).
Adicción a cocaína, anfetamina, nicotina y THC
El consumo crónico de estas sustancias produce disfun- ción de la señalización glutamatérgica y down regulation del intercambiador cistina- glutamato, así como disminución de la expresión del GLT-1 (Transportador del Glutamato Glial 1) en núcleo accumbens (NAcc). Este mecanismo estaría impli- cado en la conducta de búsqueda de sustancias, a través de la plasticidad sináptica por estimulación de receptores NMDA y del receptor metabotrópico de glutamato 5. Este aumento de la plasticidad eleva la transmisión excitatoria, relacionada con el craving (49, 50, 51). Por otro lado, ya se mencionó el efecto sobre el DAT-1, que lleva a un aumento de disponibi- lidad de DA.
Tricotilomanía, onicofagia, trastorno de excoriación, juego patológico, auto injuria no suicida (AINS)
NAC es crecientemente utilizado en conductas repetitivas mal adaptativas. Cullen et al. (52), realizaron un estudio con 18 adolescentes mujeres con AINS. Midieron la conectividad funcional en estado de reposo (RSFC por sus siglas en inglés) de la amígdala y el NAcc antes y después del tratamiento con NAC mediante neuroimagen funcional. La reducción en la frecuencia de AINS se asoció con una disminución de la RSFC en la amígdala izquierda y área motora suplementaria derecha, pero con un aumento de la RSFC en la amígdala derecha y corteza frontal inferior homolateral. En el NAcc, una reducción en la frecuencia de AINS se asoció con una disminución en la conectividad entre el NAcc derecho y la corteza frontal medial superior izquierda. También se infor- maron cambios en circuitos similares que acompañan a la mejoría clínica en la depresión y medidas de psicopatología global. Este mecanismo sería similar en otros trastornos del control de los impulsos.
TB
Está fuertemente asociado con la disfunción inmune. Es- tudios epidemiológicos replicados han demostrado que el TB tiene altas tasas de comorbilidades con enfermedades au- toinmunes e infecciosas crónicas, cardiovasculares y meta- bólicas. Los estudios de citocinas han demostrado que el TB está asociado con una inflamación crónica de bajo grado con aumentos adicionales en los niveles de citocinas proinflama- torias durante los episodios del estado de ánimo. Los meca- nismos involucrados incluyen cambios en niveles de monoa- minas inducidos por citocinas, aumento del estrés oxidativo, sobreactivación microglial patológica, sobreactivación del eje hipotálamo-pituitario suprarrenal, alteraciones del eje micro- bioma-intestino cerebral y cambios inmunitarios relacionados con el sueño (53).
EZQ
La desregulación del GSH a nivel génico, proteico y funcional conduce a la disfunción del receptor NMDA. Se encontraron niveles reducidos de GSH en LCR, cortex prefrontal y en núcleo caudado post mortem. También se detectaron defectos en su síntesis por afectación de la glutamasa cisteína ligasa. Durante el neurodesarrollo en ratas, la disminución de GSH y el aumento de DA produce síntomas similares a los que se presentan en una EZQ. La administración de antagonistas NMDA como la ketaminsa y la fenciclidina producen el mismo efecto (54, 55, 56, 57).
En un estudio cruzado, doble ciego, controlado con placebo, 19 pacientes con diagnóstico de EZQ se sometieron a dos exploraciones de resonancia magnética, luego de la administración oral de 2400 mg de NAC o un placebo equivalente. Se utilizó espectroscopía de resonancia magnética de protones para investigar el efecto de NAC en los niveles de glutamato y Glx (glutamato más glutamina) escalados a creatina (Cr) en la corteza cingulada anterior (CCA) y en el núcleo caudado derecho. Se utilizó el marcaje de espín arterial continuo pulsado para evaluar los efectos de la NAC en el flujo sanguíneo cerebral en reposo. En relación con la condición de placebo, la condición de NAC se asoció con niveles más bajos de Glx/ Cr, en la CCA (P < 0,05), pero no en el núcleo caudado. Esto es una evidencia de la regulación del glutamato por NAC en EZQ (58).
Los estudios posmortem en humanos sugieren un papel importante para las anomalías en las interneuronas GABAérgicas en la corteza prefrontal en la EZQ. Las interneuronas cortica- les diferenciadas de células madre pluripotenciales inducidas (iPSC) de sujetos con EZQ mostraron niveles significativamen- te más bajos de glutamato descarboxilasa 67 (GAD67), así como niveles reducidos de proteínas sinápticas gehpirina y NLGN2. Los cocultivos de interneuronas con neuronas piramidales corticales excitatorias de iPSC de pacientes con EZ mostraron puntos sinápticos de menor densidad y menor frecuencia del potencial de acción. La sobreexpresión de NLGN2 en dichas neuronas rescató los déficits de puntos sinápticos, mientras que la eliminación de NLGN2 en neuronas sanas
resultó en una acción contraria.
Las interneuronas también tenían un área nuclear significa- tivamente más pequeña, lo que sugiere un estado de estrés oxidativo innato.
La NAC aumentó el área nuclear en las interneuronas y la expresión de NLGN2 y rescató los déficits sinápticos. Estos resultados implican deficiencias específicas en la maquina- ria sináptica en las interneuronas corticales como reguladores críticos de las conexiones sinápticas en la EZ y apuntan a un nexo entre el estrés oxidativo y la expresión de NLGN2 en la mediación de los déficits sinápticos (59).
Conclusiones
En el trascurso de los últimos años se han incrementado las evidencias sobre los mecanismos inmunitarios e inflamatorios involucrados en la fisiopatología de las enfermedades neurop- siquiátricas, lo cual contribuyó a la búsqueda de moléculas que actúan modulándolos, con el objetivo de mejorar las ma- nifestaciones de las entidades mencionadas. Dentro de ellas, se encuentran fármacos utilizados previamente en trastornos clínicos y cardiovasculares, como la NAC, los ácidos omega 3 y 6 de cadena larga y los AINE, entre otros.
En el caso de la NAC, son promisorios los resultados ob- tenidos a la fecha, sumado a su favorable perfil de efectos adversos y costo. No obstante, se necesitan más ensayos con- trolados involucrando cohortes más amplias para establecer conclusiones firmes.
Referencias bibliográficas
- 1. Aruoma, O, Halliwell B, Hoey B, Butler J. 1989. The antioxidant action of N acetylcystei- ne: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radical Biol. Med. 6: 593–597.
- 2. Bavarsad Shahripour R, Harrigan M, Alexan- drov A (2014). N-acetylcysteine (NAC) in neu- rological disorders: mechanisms of action and therapeutic opportunities. Brain and Behavior; 4(2): 108–122.
- 3. Dekhuijzen, P. 2004. Antioxidant properties of N-acetylcysteine: their relevance in relation to chronic obstructive pulmonary disease. Eur. Respir. J. 23: 629–636.
- 4. Ziment I (1988). Acetylcysteine: a drug that is much more than a mucokinetic. Biomed Phar- macother;42(8): 513-9.
- 5. Ramos-Villegas Y, Padilla-Zambrano H, Blanco-Teherán C, López-Cepeda D, Quinta- na-Pájaro L, Corrales-Santander H et al. (2017). N-Acetilcisteína en neuroprotección y lesión traumática cerebral: revisión de la literatura. Rev. Chil. Neurocirugía 43: 166-169.
- 6. Sen, C. (1997). Nutritional biochemistry of celular glutathione. J. Nutr. Biochem. 8: 660–672.
- 7. Samuni, Y, Goldstein S, Dean O, Berk M (2013). The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 1830: 4117–4129.
- 8. Kerksick, C, Willoughby D (2005). The an- tioxidant role of glutathione and N-acetyl-cystei- ne supplements and exercise-induced oxidative stress. J. Int. Soc. Sports Nutr. 2 :38–44.
- 9. Meister A. (1995) Glutathione metabolism. Methods Enzymol. 251 :3.
- 10. Xu Y, Hou X, Liu Y, Zong Y (2009). Di- fferent protection of K252a and N-acetyl-L-cys- teine against amyloid-beta peptide-induced
cortical neuronapoptosis involving inhibition of MLK3-MKK7-JNK3 signal cascades. J Neorosci Res, 87(4): 918-27, 2009 Mar.
- 11. Tian H, Zhang Q, Li H, Zhang G (2003). Antio xidant N-acetylcysteine and AMPA/KA re- ceptor antagonist DNQX inhibited mixed lineage kinase-3 activation following cerebral ischemia in rat hippocampus. Neurosci Res; 47(1): 47- 53.
- 12. Su B, Mitra S, Gregg H, Flavahan, Cho- tani S, Clark K, et al. (2001). Redox regulation of vascular smooth muscle cell differentiation. Circ. Res. 89:39–46.
- 13. Ichiki T, Takeda K, Tokunou T, Funakos- hi Y, Ito K, Iino N, et al. 2001. Hypertension 37:535–540.
- 14. Ghigliotti G, Mereto E, Eisenberg P, Mar- telli A, Ors P, Sini D, et al. (2001). N-acetyl-cys- teine reduces neointimal thickening and procoa- gulant activity after balloon-induced injury in abdominal aortae of New Zealand white rabbits. Thromb. Haemost. 85:724–729.
- 15. Yan Z, Subbaramaiah K, Camilli T, Zhang F, Tanabe T, McCaffrey T, et al. (2000). Ben- zo[a]pyrene induces the transcription of cy- clooxygenase-2 in vascular smooth muscle cells. Evidence for the involvement of extracelular signal-regulated kinase and NF-kappaB. J. Biol. Chem. 275: 4949–4955.
- 16. Nagase M, Ando K, Nagase T, Kaname S, Sawamura T, Fujita T. (2001). Redox-sensitive regulation of lox-1 gene expression in vascular endothelium. Biochem. Biophys. Res. Commun. 281: 720–725.
- 17. Mass H, Pirazzi B, Gonzalez P, Collazo V, Fitzovich D, Avakian E. (1995). N-acetylcystei- ne diminishes injury induced by balloon angio- plasty of the carotid artery in rabbits. Biochem. Biophys. Res. Commun. 215: 613–618.
- 18. De Mattia, G, Bravi M, Laurenti O, Cas- sone-Faldetta M, Proietti M, De Luca O et al.
(1998). Reduction of oxidative stress by oral N-acetyl-L-cysteine treatment decreases plasma soluble vascular cell adhesion molecule-1 con- centrations in non-obese, non-dyslipidaemic, normotensive, patients with non-insulin-depen- dent diabetes. Diabetologia 41: 1392–1396.
- 19. Martin K, Kari F, Barrett J, French J. (2000). N-acetyl-L-cysteine simultaneously in- creases mitogenesis and suppresses apoptosis in mitogen-stimulated B-lymphocytes from p53 haploinsufficient Tg. AC (v-Ha-ras) mice. In Vitr. Mol. Toxicol. 13: 237–247.
- 20. Galle J, Heermeier K, Wanner C. (1999). Atherogenic lipoproteins, oxidative stress, and cell death. Kidney Int.56:S62–S65.
- 21. Tyagi S (1998). Homocysteine redox re- ceptor and regulation of extracellular matrix components in vascular cells. Am. J. Physiol. 274: C396–C405.
- 22. Sarker K, Abeyama K, Nishi J, Nakata M, Tokioka T, Nakajima N et al. 1999. Inhibition of thrombin-induced neuronal cell death by recom- binant thrombomodulin and E5510, a synthetic thrombin receptor signaling inhibitor. Thromb. Haemost. 82: 1071–1077.
- 23. Silva Rodrigues F, Patrícia Françae A, Broettoa N, Flávia Furianf A, Schneider Oliveira M, Soares Santos A et al (2020). Sustained glial reactivity induced by glutaric acid may be the trigger to learning delay in early and late phases of development: Involvement of p75NTR recep- tor and protection by N-acetylcysteine. Brain Research Vol 1749, 15, 147145
- 24. Abello P, Fidler S, Buchman T (1994). Thiol reducing agents modulate induced apopto- sis in porcine endothelial cells. Shock 2: 79–83.
- 25. Hong D, Kho A, Lee S, Jeong J, Kang B, Kang D et al. (2020). Transient Receptor Po- tential Melastatin 2 (TRPM2) Inhibition by An- tioxidant, N-Acetyl-l-Cysteine, Reduces Global Cerebral Ischemia-Induced Neuronal Death. Int.
J. Mol. Sci. 2020, 21, 6026; doi: 10.3390 /
ijms21176026.
- 26. Jiang L, Yang W, Zou J, Beech D (2010). TRPM2 channel properties, functions and the- rapeutic potentials. Expert Opin. Ther. Targets 14, 973–988.
- 27. Unnithan A, Choi H, Titler A, Posimo J, Leak R. (2012). Rescue from a two hit, hi- gh-throughput model of neurodegeneration with N-acetyl cysteine. Neurochem. Int. 61: 356–
368.
- 28. Arakawa M, Ito Y (2007). N-acetylcysteine and neurodegenerative diseases: basic and clini- cal pharmacology. Cerebellum 6: 308–314.
- 29. Devi P, Pillai K, Vohora D (2006). Faci- litation Action of N-Acetylcysteine on the Anti- convulsant Effect of Sodium Valproate in Mice. Basic & Clinical Pharmacology & Toxicology.98, 521–522.
- 30. Behar T, Colton C (2003). Redox regula- tion of neuronal migration in a Down Syndrome model. Free Radical Biol. Med. 35: 566–575.
- 31. Padilha Marchetti D, Donida B, Deon M, Jacques C, Bannach Jardim L, Regla Vargas C (2007). In vitro effect of N-acetyl-L-cysteine on glutathione and sulfhydryl levels in X-linked adrenoleukodystrophy patients. Clin Biomed Res;37(1): 33-37.
- 32. Engelen M, Kemp S, de Visser M, Geel B, Wanders R, Auboourg P. et al. X-linked adre- noleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and ma- nagement (2012). Orphanet J Rare Dis 7, 51.
- 33. Kartha R, Zhou J, Basso L, Schröder H, Or- chard P, Cloyd J (2015). Mechanisms of Antioxi- dant Induction with High-Dose N-Acetylcysteine in Childhood Cerebral Adrenoleukodystrophy. CNS Drugs, 29(12), 1041-1047.
- 34. Sadan O, M. Bahat-Stromza, Y. Gil- gun-Sherki, D. Atlas, E. Melamed, Offen D (2005). A novel brain-targeted antioxidant (AD4) attenuates haloperidol-induced abnormal movement in rats: implications for tardive dyski- nesia. Clin. Neuropharmacol. 28: 285–288
- 35. Fontaine M, Geddes J, Banks A, Butter- field D (2000). Effect of exogenous and endo- genous antioxidants on 3-nitropionic acid-indu- cedin vivo oxidative stress and striatal lesions. J. Neurochem. 75: 1709–1715.
- 36. Sandhir R, Sood A, Mehrotra A, Kamboj S (2012). N-Acetylcysteine reverses mitochon- drial dysfunctions and behavioral abnormalities in 3-nitropropionic acid-induced Huntington’s disease. Neurodegener Dis; 9: 145–157.
- 37. Clark J, Clore E, Zheng K, Adame A, Mas- liah E, Simon D. (2010). Oral N-acetyl-cysteine attenuates loss of dopaminergic terminals in al- pha-synuclein overexpressing mice. PLoS ONE 5: e12333.
- 38. Monti D, Zabrecky G, Kremens D, Liang T, Wintering N, Cai J et al. (2016). N-Acetyl Cys- teine May Support Dopamine Neurons in Parkin- son’s Disease: Preliminary Clinical and Cell Line Data. PLoS One Jun 16;11(6): e0157602.
- 39. Erikson M, Hansen K, Banks W (2012). In- flammation-induced dysfunction of the low-den- sity lipoprotein receptor-related protein-1 at the blood-brainbarrier: protection by the antioxidant N-acetylcysteine. Brain Behav Immun; 26(7): 1085-94.
- 40. Lehmann D, Karussis D, Misrachi-Koll R, Shezen E, Ovadia H, Abramsky O. (1994). Oral administration of the oxidant-scavenger N-ace- tyl-L-cysteine inhibits acute experimental au- toimmune encephalomyelitis. J. Neuroimmunol. 50: 35–42.
- 41. Rosen D, Siddique T, Patterson D, Fi- glewicz D, Sapp P, Hentati A, et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362: 59–62.
- 42. Vasdekis S, Athanasiadis D, Lazaris A, Martikos G, Katsanos A, Tsivgoulis G (2013). The role of remote ischemic preconditioning in the treatment of atherosclerotic diseases. Brain Behav. 3: 606–616.
- 43. Morris K, H. W. Lin H, Thompson J, Pe- rez-Pinzon A (2011). Pathways for ischemic cytoprotection: role of sirtuins in caloric restric- tion, resveratrol, and ischemic preconditioning.
J. Cereb. Blood Flow Metab. 31: 1003–1019.
- 44. Sury M, Frese- Schaper M, Mühlemann M, Schultess F, Blasig I, Ingolf E et al. (2006). Evidence that N-acetylcysteine inhibits TNF-al- pha-induced cerebrovascular endothelin-1 upre- gulation via inhibition of mitogen- and stress-ac- tivated protein kinase. Free Radic Biol Med; 41(9): 1372-83.
- 45. Sharabi H, Khatib N, Ginsberg, Y, Weiner Z, Ross M, Tamar B et al. (2019). Therapeutic N-Acetyl-Cysteine (Nac) Following Initiation of Maternal Inflammation Attenuates Long-Term Off spring Cerebral Injury, as Evident in Mag- netic Resonance Imaging (MRI). Neuroscience; 403: 118-124.
- 46. Simpson E, Kellendonk C, Kandel E (2010). A Possible Role for the Striatum in the Pathogenesis of the Cognitive Symptoms of Schizophrenia. Neuron 65, 585–596.
- 47. Di Pietro P, Dias M, Scaini G, Burigo M, Constantino L, Machado R et al. (2008). Inhi- bition of brain creatine kinase activity aft er re- nal ischemia is attenuated by N-acetylcysteine and deferoxamine administration. Neurosci Lett; 434(1): 139-43.
- 48. Kamboj S, Sandhir R (2011). Protective effect of N-acetylcysteine supplementation on mitochondrial oxidative stress and mitochon- drial enzymesin cerebral cortex of streptozoto- cin-treated diabetic rats. Mitochondrion; 11(1): 214-22.
- 49. Gipson C, Reissner K, Kupchik Y, Smith A, Stankeviciute N, Hensley-Simon M et al. (2013) Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc Natl Acad Sci U S A;110(22): 9124–9.
- 50. Baker D, McFarland K, Lake R, Shen H, Toda T, Kalivas P (2003). N-acetyl cysteine-in-
duced blockade of cocaine-induced reinstate- ment. Ann N Y Acad Sci.;1003: 349–51.
- 51. Palmatier M, Liu X, Donny E, Caggiula A, Sved A (2008). Metabotropic glutamate 5 receptor (mGluR5) antagonists decrease nicoti- ne seeking, but do not affect the reinforcement enhancing effects of nicotine. Neuropsychophar- macology, 33(9): 2139–47.
- 52. Cullen K, Schreiner M, Klimes-Dougan B, Eberly L, LaRiviere L, Lim K et al. (2020). Neural correlates of clinical improvement in res- ponse to N-acetylcysteine in adolescents with non-suicidal self-injury. Prog Neuropsychophar- macol Biol Psychiatry; 99: 109778.
- 53. Rosenblat J, McIntyre R (2017). Bipolar Disorder and Immune Dysfunction: Epidemio- logical Findings, Proposed Pathophysiology and Clinical Implications. Brain Sci. 2017, 7, 144; doi:10.3390/brainsci7110144.
- 54. Lavoie S, Murray M, Patricia Deppen P, Knyazeva M, Berk M, Boulat O et al. (2008). Glutathione Precursor, N-Acetyl-Cysteine, Im- proves Mismatch Negativity in Schizophrenia Patients. Neuropsychopharmacology 33, 2187–
2199.
- 55. Gysin R, Kraftsik R, Sandell J, Bovet P, Chappuis C, Conus P et al. (2007). Impaired glutathione synthesis in schizophrenia conver- gent genetic and functional evidence. Proc Natl Acad Sci USA 104: 16621–16626.
- 56. Robbins T (2005). Synthesizing schi- zophrenia: a bottom-up, symptomatic approach. Schizophr Bull 31: 854–864.
- 57. Krystal J, Karper L, Seibyl J, Freeman G, Delaney R, Bremner J et al. (1994). Subanes- thetic effects of the noncompetitive NMDA anta- gonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine res- ponses. Arch Gen Psychiatry 51: 199–214.
- 58. McQueen G, Lally J, Collier T, Zelaya F, Lythgoe D, Barker D et al. (2018). Effects of N-acetylcysteine on brain glutamate levels and resting perfusion in schizophrenia. Psychophar- macology (Ber) 235(10): 3045-3054.
- 59. Kathuria A, Lopez-Lengowski K, Watmuff B, McPhie D, Cohen B, Karmacharya R. (2019) Synaptic deficits in iPSC-derived cortical in- terneurons in schizophrenia are mediated by NLGN2 and rescued by N-acetylcysteine. Trans- lational Psychiatry 9: 321.





No comments! Be the first commenter?