

Farmacotecnia en neuropsicofarmacología Los sistemas de liberación modificada de psicofármacos en la práctica clínica cotidiana
Resumen
La farmacología es la ciencia biológica que estudia las acciones y propiedades de los fármacos en los organismos, siendo un fármaco toda sustancia química utilizada en el tratamiento, curación, prevención o diagnóstico de una enfermedad o, en su de- fecto, para evitar la aparición de un proceso fisiológico no deseado. Todo medicamento, cualquiera sea su vía de administración, cumple necesariamente con una fase farmacéutica, una fase farmacocinética y, finalmente, una fase farmacodinámica. Se estudiará en este resumen, las características cinéticas diferenciales en torno a los distintos sistemas de liberación modificada, sus pros y sus contras para la dinámica diaria.
Palabras clave
Farmacología – Liberación extendida – Liberación controlada – Farmacocinética.
Muñoz S, Mejías Delamano A, Serra HA, Müller F. “Farmacotecnia en neuropsicofarmacología. Los sistemas de liberación modificada de psicofármacos en la práctica clínica cotidiana. Parte I”. Psicofarmacología 2023;132:14-25.
Introducción
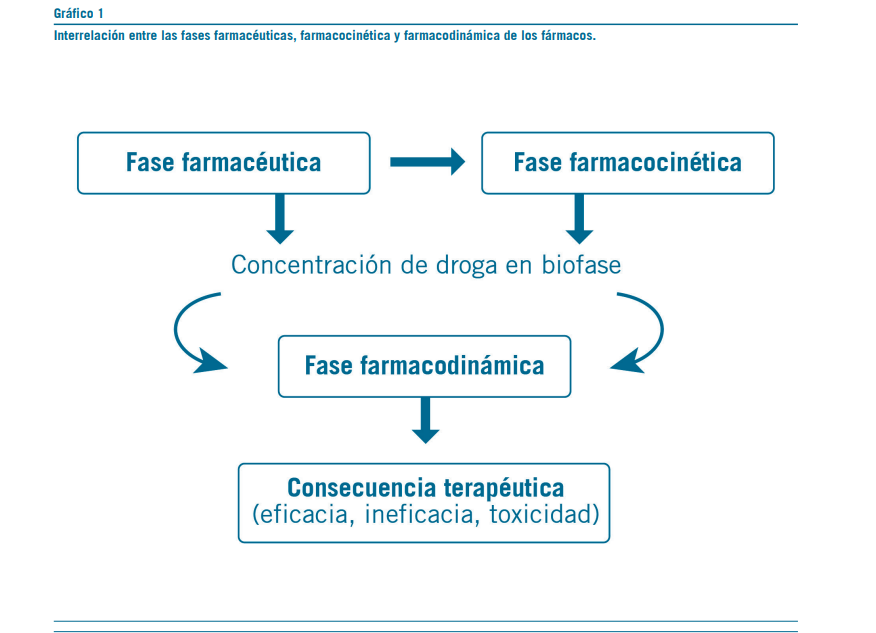
La farmacología es la ciencia biológica que estudia las acciones y propiedades de los fármacos en los organismos, siendo un fármaco toda sustancia química utilizada en el tratamiento, curación, prevención o diagnóstico de una enferme- dad o, en su defecto, para evitar la aparición de un proceso fisiológico no deseado. Por otro lado, la farmacocinética estudia todos aquellos procesos y factores que determinan la cantidad de fármaco presente en el sitio en que debe ejercer su efecto biológico (denominado biofase) en cada momento, a partir de la aplicación del fármaco sobre el organismo vivo (administración). La farmacocinética es un proceso dinámico, donde todos los procesos intervinientes ocurren simultánea- mente. Asimismo, la farmacodinamia estudia las acciones y los efectos de los fármacos sobre un organismo. Todo medicamento, cualquiera sea su vía de administración, cumple necesariamente con una fase farmacéutica, una fase farmacocinética y, finalmente, una fase farmacodinámica (Gráfico 1).
Aspectos farmacocinéticos
La vía de administración es el sitio donde se aplica el me- dicamento para que pueda ser absorbido o, en su defecto, que actué en dicho sitio. Existen 2 tipos generales de vías de administración de fármacos:
- vías enterales: oral, sublingual, rectal
- vías parenterales: intravenosa, intraarterial, intramuscu- lar, subcutánea.
La vía de administración entérica utiliza las mucosas bucal, gástrica, intestinal y rectal como sitios de absorción de las moléculas de un fármaco. La vía de administración oral es la más fisiológica y cómoda, depende de la colaboración del paciente, puede generar irregularidades de las concentracio- nes plasmáticas cuando existen alteraciones del peristaltismo
normal o vómitos y puede irritar el tracto gastrointestinal. Asi- mismo, esta vía de administración se encuentra influenciada por el fenómeno de metabolismo de primer paso hepático ya que la absorción se lleva a cabo en el estomago o el intestino delgado. Además, existen otras vías de administración de los fármacos (inhalatoria, dérmica, nasal, etc.) las cuales pre- sentan ventajas y desventajas que condicionan su elección. Además, los fármacos presentan determinadas vías de admi- nistración de acuerdo con su tipo de absorción, propiedades fisicoquímicas y sus preparaciones farmacéuticas.
Todos los procesos farmacocinéticos (absorción, distribu- ción, metabolismo y eliminación – ADME) requieren el pasa- je de las moléculas del fármaco a través de las membranas biológicas formadas por una doble capa lipídica en la que se intercalan diferentes tipos de proteínas. Las moléculas de pequeño tamaño atraviesan las membranas por medio de la
difusión pasiva, la difusión facilitada o por medio del trans- porte activo. Las moléculas de gran tamaño lo hacen a través de procesos de pinocitosis y exocitosis. La velocidad de difu- sión a través de la bicapa lipídica depende del tamaño de la molécula, su liposolubilidad y su grado de ionización:
- las moléculas pequeñas y no polares difunden a través de las membranas biológicas con mayor rapidez.
- las moléculas polares sin carga eléctrica difunden con rapidez si son pequeñas y, con lentitud, si son polares.
- las moléculas ionizadas, por pequeñas que sean, no atraviesan fácilmente la barrera lipídica.
La difusión simple es el mecanismo de transporte de fármacos

más frecuentemente utilizado por las moléculas de las drogas. La mayor parte de los fármacos tienen un tamaño pequeño/ mediano que permite su pasaje a través de las membranas bio- lógicas por difusión a favor de un gradiente de concentración (cuando no se encuentran ionizados). La velocidad del pasaje de las drogas, según la Ley de Fick, será mayor cuanto mayor sea el gradiente de concentración del fármaco, cuando el tamaño de la molécula sea menor y su liposolubilidad sea mayor. A su vez, la liposolubilidad depende del grado de ionización: la forma ionizada difunde en forma dificultosa a través de la membrana plasmática, mientras que la forma no ionizada di- fundirá fácilmente hasta que se equilibre la concentración de

la droga a ambos lados de la membrana biológica.
La absorción es el traslado de las moléculas de un fárma- co desde el sitio de administración hasta el compartimiento central. En el caso de las formas farmacéuticas sólidas (com- primidos, cápsulas), la absorción requiere inicialmente la di- solución del comprimido o cápsula, liberando de esta manera el principio activo. En términos generales, las drogas se absor- ben a lo largo del tracto gastrointestinal: en primera instancia en el estómago con un pH menor a 4 donde se absorben las drogas (ácidos débiles) y luego en el intestino delgado donde el pH es mayor a 6 y se absorben mejor las formas básicas de los fármacos. Es importante recordar que la superficie de absorción del intestino es mucho más grande e irregular que la del estó-
mago teniendo una superficie de contacto mucho más extensa. Se define como liberación al proceso mediante el cual un principio activo (fármaco) presente en una forma de dosificación determinada llega a estar disponible para su absorción. En las formas farmacéuticas sólidas, en las que el medicamento no se encuentra de antemano disuelto (comprimidos, cápsulas, tabletas), el proceso de liberación comprende la desintegración del mismo. La liberación implica la disolución del principio activo en los fluidos corporales. Por lo tanto, el fármaco debe estar en condiciones adecuadas para su absorción (en su forma libre, no asociado con ninguna macromolécula como es el caso de las proteínas plasmáticas).
En aquellos fármacos en los que el efecto terapéutico depende directamente de la concentración alcanzada en la bio-

fase y en los que esta concentración se encuentra en equilibrio con la concentración sérica (plasmática), se puede establecer una relación entre el curso temporal de las concentraciones plasmáticas y los efectos terapéuticos observados. Se utilizan diversos parámetros farmacocinéticos para determinar el efecto de los fármacos:
- Concentración Mínima Eficaz (CME): es la concentración sérica por encima de la cual se observa el efecto terapéutico del fármaco.
- Concentración Mínima Tóxica (CMT): es aquella concentración de una droga por encima de la cual se observan efectos tóxicos. El índice terapéutico (IT) consiste en el cociente entre la CMT y la CME, mientras mayor sea este índice terapéutico, mas fácil será conseguir efectos terapéuticos sin que se desarrollen efectos tóxicos.
- Periodo de Latencia (PL): es el tiempo que transcurre desde la administración del fármaco hasta el comienzo del efecto farmacológico, o sea, hasta que alcanza la CME.
- Intensidad del efecto (IE): para algunos fármacos se relaciona con la concentración máxima que se alcanza, pero la concentración en los tejidos puede variar debido a la unión a las proteínas plasmáticas, el flujo sanguíneo regional o la afinidad del fármaco por los receptores.

- Duración de la acción: es el tiempo transcurrido entre el momento en el que se alcanza el CME y el momento en que desciende por debajo de esta, durante el cual se evidencian los efectos terapéuticos del fármaco.
Dentro de la Neuropsicofarmacología, existen diversos fár- macos que requieren del establecimiento de un rango tera- péutico para determinar si el paciente posee concentraciones plasmáticas que generen un efecto terapéutico adecuado y no estén en peligro de un desarrollo de reacciones adversas medicamentosos (RAM). Algunos de los principios activos que necesitan una determinación del rango terapéutico son el li- tio, ácido valproico y la clozapina.
Farmacotecnia
Desde hace años la tecnología farmacéutica implementa distintas modificaciones a las formas farmacéuticas tradicio- nales de los medicamentos que permiten cambiar o retrasar el sitio de absorción del o los principios activos para generar beneficios farmacocinéticos. Básicamente, el objetivo princi- pal es lograr niveles plasmáticos adecuados de la droga en el órgano diana (target). La farmacotecnia es el estudio y aplica-
ción de cambios intencionales en la formulación de los medi- camentos con el objetivo de modificar la velocidad, el lugar o momento de la liberación del principio activo en el organismo humano, lo que normalmente se conoce como forma farma- céutica de liberación modificada (FFLM).
La Farmacopea Norteamericana (USP – U.S. Pharmaco- peian) define a las formas farmacéuticas de liberación modi- ficada (FFLM) como aquellas formulaciones en las cuales se eligen características de liberación en el curso del tiempo y/o localización para lograr objetivos terapéuticos o de convenien- cia que no ofrecen las formas farmacéuticas tradicionales. Por otro lado, la autoridad sanitaria de Estados Unidos, la Food and Drug Administration (FDA) utiliza el término “liberación controlada” para referirse a las FFLM. Cabe destacar que las formas farmacéuticas de liberación controladas se diferencian en aquellas de liberación extendida o sostenida y de otras de liberación retardada. Finalmente, la Real Farmacopea Es- pañola se refiere a un sistema de liberación modificada de drogas cuando se cambia la velocidad, el lugar o el momento de liberación del principio activo una vez que se administra la forma farmacéutica.
Las FFLM presentan diversos beneficios y desventajas en la práctica clínica. Dentro de los beneficios se destacan la mayor
aceptación y adherencia terapéutica de los pacientes, mejor comodidad posológica, un efecto terapéutico prolongado, dis- minución de la fluctuación de los niveles plasmáticos y la reducción de los efectos adversos relacionados con los picos plasmáticos. Por otro lado, las desventajas incluyen: requeri- miento de no alterar las características de la formulación (par- tir o triturar un comprimido), pérdida de la flexibilidad en el ajuste de dosis, la variabilidad interindividual en la absorción de la droga y un mayor precio de la especialidad medicinal (Ver Cuadro 1).
Con este tipo de medicamentos se intenta lograr uno de los siguientes objetivos:
- lograr con rapidez una concentración plasmática del fár- maco que se mantenga relativamente constante y dentro de la ventana terapéutica durante un período de tiempo suficiente.
- conseguir rápidamente una concentración plasmática del fármaco que, aunque no permanezca constante, disminuya lo suficientemente despacio como para mantenerse dentro de la ventana terapéutica durante un período de tiempo suficiente.
Las formulaciones de liberación modificada utilizan una “barrera” química o física para hacer más lenta la liberación de la dosis de mantenimiento del fármaco. Entre ellas están el uso de revestimientos, la inclusión del fármaco en un matriz de cera o plástico, la microencapsulación, la fijación quími- ca a resinas de intercambio iónico y la incorporación de una bomba osmótica. Los medicamentos que se benefician más con la formulación de liberación modificada son aquellos que tienen alguna de las siguientes características:
- estrecho margen terapéutico (teofilina, litio)
- efectos adversos relacionados con la concentración plas-
mática (nifedipina, verapamilo, diltiazem)
- duración de acción corta que requiere 3 o más administra- ciones diarias (morfina, tramadol, pramipexol)
- una breve vida media que requiere la administración en 2 o 3 tomas diarias (pregabalina, levetiracetam)
- tolerancia farmacológica con niveles plasmáticos constan- tes (nitratos).
A continuación, se desarrollarán los distintos tipos de siste- mas de liberación modificada de fármacos.
Liberación retardada, retrasada o diferida
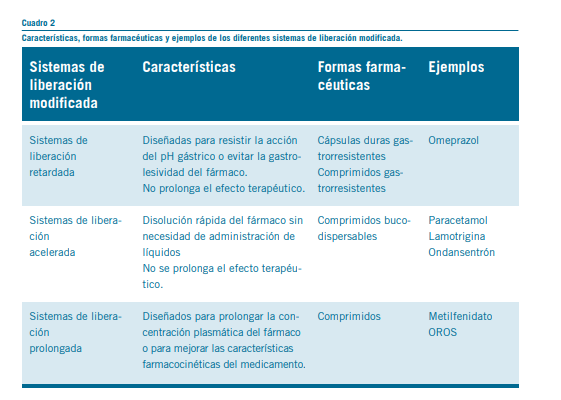
Es la formulación que requiere para el inicio de la absorción del principio activo el transcurso de un determinado periodo de tiempo después de la administración. Constituye una forma farmacéutica con una cubierta entérica (también llamada sensible al pH), en la que el principio activo se libera en una zona particular del intestino delgado, pasando por el estomago sin degradarse. Este tipo de forma farmacéutica protege al principio activo de la acción nociva del ácido estomacal, especialmente aquellos que son sensibles al mismo, evitando su degradación y afectando su normal absorción. El principio activo es liberado en un momento diferente al de la administración y no prolonga el efecto terapéutico del fármaco, ya que no existe otro cambio farmacocinético. La cubierta entérica es generalmente un polímero que protege al comprimido del medio ácido. Asimismo, es adecuada para envolver gránulos, comprimidos o macropartículas. Algunos ejemplos de formu- laciones de liberación retardada son:
- Comprimidos gastrorresistentes
- Cápsulas gastrorresistentes
- Sistemas colónicos (sistemas de liberación de fármacos en la primera porción del colon).
En definitiva, la formulación de liberación retardada/retrasada incluye a todas las formas farmacéuticas que contienen
una cubierta entérica cuyo principal objetivo es evitar la acción erosiva de determinadas drogas sobre la mucosa del estómago y, de esta manera, evitar lesionar el equilibrio fisiológico de la misma. Esto es válido para drogas ácidas (por ejemplo, algunos analgésicos como el ácido acetilsalícilico, ibuprofeno y diclofe- nac) y otros fármacos no ácidos (por ejemplo, el inhibidor de la bomba de protones omeprazol que se degrada al ser some- tido a un medio ácido) para asegurar su absorción. Asimismo, permiten una mejor posología del tratamiento y minimizan los efectos adversos gastrointestinales. Es importante destacar que no aumentan el efecto terapéutico del fármaco y desde un pun- to de vista farmacocinético, sólo se observa un desplazamiento
de la curva concentración plasmática/tiempo hacia la derecha.
Sistemas de liberación acelerada
En este sistema de liberación de fármacos, el principio ac- tivo se disuelve en forma instantánea en la cavidad bucal sin requerir la administración de agua o líquidos. El medicamento en contacto con la saliva se desintegra rápidamente por la ayuda de los excipientes que tiene en su composición. Los tipos de presentaciones de liberación acelerada son:
- Comprimidos efervescentes en contacto con la saliva (por ejemplo, paracetamol)

2) Tabletas liofilizadas: son matrices que contienen el principio activo que ante el contacto con cualquier solución acuosa se desintegran rápidamente. Algunos ejemplos son el antiinflamatorio no esteroide piroxicam, la olanzapina y mir- tazapina.
Conceptualmente, el principio activo se libera rápidamen- te, aunque no necesariamente significa que su absorción sea más rápida. La diferencia radica en que el principio activo se libera o dispersa rápidamente sin necesidad de administra- ción de agua, pero no existe diferencia en el inicio de acción farmacológica.
Sistemas de liberación controlada
En este sistema de liberación, el principio activo se va li- berando gradualmente con el tiempo a una velocidad limita- da por el sistema de liberación y tiene como consecuencia la prolongación del efecto terapéutico. Cabe destacar que la velocidad de liberación del principio activo es limitante en el proceso farmacocinético de absorción. Las formas farmacéu- ticas de liberación prolongada incluyen: forma de liberación sostenida o continua y la forma de liberación prolongada (LP)
- Formas de liberación sostenida o continuada: permiten que el fármaco se libere a una velocidad constante, logrando así una velocidad de absorción de la droga en forma constante y, finalmente, disminuyendo la fluctuación de los niveles séricos del medicamento. Algunos ejemplos de fármacos con este sis- tema de liberación es la nifedipina y el metilfenidato OROS. El sistema OROS (también denominada bombas osmóticas o sistema GITS) se basa en la integración del principio activo y el sistema osmótico en una membrana semipermeable. Se realiza la introducción de un agente osmótico y el fármaco en una membrana semipermeable. Cuando el agua penetra, el principio activo disuelto se libera en forma controlada a través de un orificio pequeño realizado con un laser. Este sistema consta de una triple capa: una matriz, el principio activo y una membrana. La capa inferior es una matriz osmótica por encima de la cual se encuentra la droga y luego un recubri- miento polimérico. La droga es expulsada a través de un micro orificio logrado con tecnología laser e impulsado a través de un gradiente osmótico posterior al ingreso de agua. De esta manera se logra una liberación constante del principio activo a través del tiempo.
- Formas de liberación prolongada: este sistema de libera- ción (también denominada liberación extendida (XR / ER – extended release) es un tipo de liberación que es lo suficien- temente lenta para poder duplicar el intervalo de dosificación y aun más. Esta formulación no libera el principio activo in- mediatamente después de su administración y permite una reducción en la frecuencia de dosificación. Permiten que se logre un aumento significativo en el cumplimiento de la toma del medicamento por parte del paciente o una mejoría im- portante en el desempeño terapéutico del medicamento. Se
basa por medio de dispersiones del fármaco en un sistema de matriz polimérico que puede ser hidrofílico o lipofílico se puede lograr la liberación regulada del principio activo. Esta tecnología es utilizada para las formulaciones de morfina de liberación prolongada MST
A continuación, se resumen las principales características de los diferentes sistemas de liberación modificada de fárma- cos (Ver Cuadro 2).
Cabe destacar que las características de liberación modifi- cada de las especialidades medicinales con estas caracterís- ticas se pierden al dividir, partir, triturar o masticar los com- primidos ya que estos preparados están diseñados para ser tragados enteros. La subdivisión puede provocar la liberación inmediata de cantidades tóxicas de fármaco alterando su bio- disponibilidad y, de acuerdo del grupo farmacológico, puede comprometer la vida del paciente.
Existen 2 tipos de formulaciones de liberación modificada con características particulares: las formulaciones sublingua- les y los parches transdérmicos.
Formas farmacéuticas sublinguales
La mucosa sublingual ofrece una superficie de absorción pequeña. Esta mucosa es permeable al pasaje de drogas no ionizadas y muy liposolubles. Por estos motivos, solamente se pueden administrar por esta vía aquellos fármacos lo sufi- cientemente potentes para que se logre un efecto terapéutico a pesar del pasaje de escasas moléculas de la droga a la cir- culación sistémica.
Las formas farmacéuticas sublinguales (SL) generalmente son de forma lenticular y pequeñas (de esta manera presen- tan una mejor superficie de absorción) y deben ser colocadas bajo la lengua. Esta vía está limitada por el peso molecular, grado de ionización, pH y coeficiente de liposolubilidad del fármaco. La forma sublingual se encuentra reservada solo a un pequeño número de principios activos (por ejemplo, clo- nazepam, lorazepam, alprazolam, zolpidem). Su efecto tera- péutico es bastante rápido y la absorción directa a través de la mucosa sublingual que permite la llegada de la droga al torrente sanguíneo con la ventaja de evitar el metabolismo de primer paso hepático. La desventaja principal es que está re- servada solamente para aquellos medicamentos cuya dosis es pequeña y cuyo principio activo pueda absolverse por esta vía.
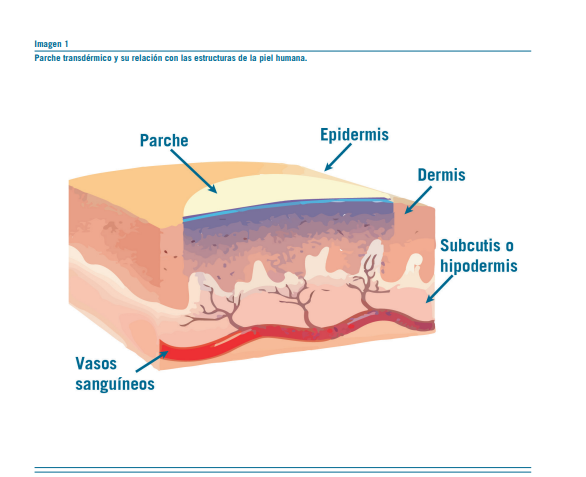
Parches transdérmicos
Los parches transdérmicos son formas farmacéuticas de li- beración continua que se aplican directamente sobre la piel. Su objetivo es lograr un efecto sistémico por medio de un sistema transdérmico, también conocido como TTS en sus siglas en inglés (transdermal therapeutic systems). Los sis- temas transdérmicos permiten controlar la posología y soste- ner durante el tiempo los niveles plasmáticos constantes del fármaco durante el periodo de aplicación del parche. Es de utilidad para aquellos fármacos utilizados en el tratamiento
prolongado y que requieren bajos niveles séricos que se deben mantener constantes durante extensos periodos de tiempo. Algunas patologías que aplican estos criterios son la angina de pecho, patologías hormonales, la anticoncepción, las patologías dolorosas y la demencia tipo Alzheimer.
El parche transdérmico está compuesto por un principio activo inmerso en un polímero tridimensional con una capa protectora oclusiva y una capa adhesiva superficial. En general, la matriz o una membrana con una superficie determinada permiten controlar el paso de las moléculas del fármaco a una velocidad y cantidad constante a través del tiempo. Una de las precauciones importantes es que no se debe cortar el parche porque se pueden alterar sus características. Algunos parches poseen un coadyuvante que favorece la absorción. Una de las ventajas farmacocinéticas del parche transdérmico es que las moléculas de la droga llegan al torrente circulatorio evitando el primer paso hepático.
Esta vía de administración está limitada a aquellos principios activos con un determinado coeficiente de partición lípido/agua y peso molecular de modo que pueda atravesar el estrato córneo y no quede excesivamente atrapado. Estos sistemas en general tienen características estéticas, hipoaler- génicas y de adaptabilidad para lograr su objetivo terapéutico.
Algunos ejemplos de fármacos que poseen presentaciones en parches transdérmicos son: rivastigmina, fentanilo, buprenor- fina, estradiol, entre otros (Ver Imagen 1).
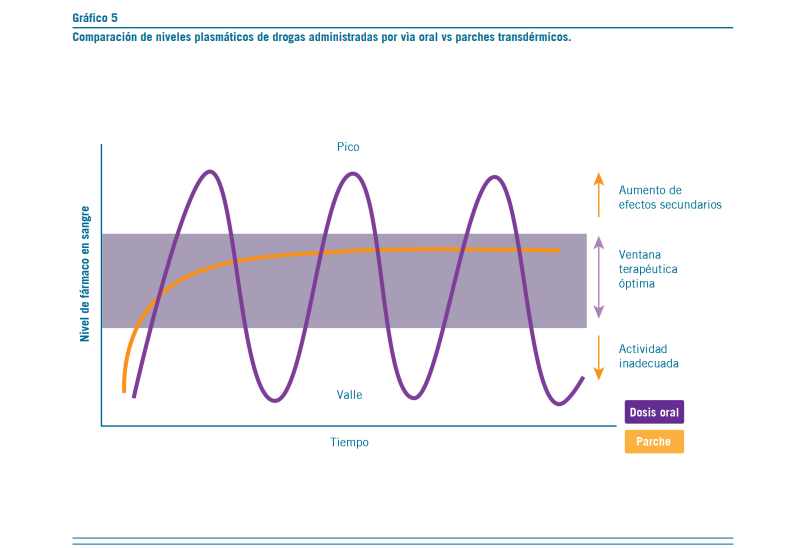
Los niveles plasmáticos que facilitan los parches transdérmicos son diferentes a los obtenidos con las formulaciones orales del mismo fármaco (Ver Gráfico 5).
Las ventajas del parche transdérmicos se resumen en el cuadro 3 (Ver Cuadro 3).
En resumen, los sistemas de liberación modificada constituyen una opción de administración de fármacos con determinadas características fisicoquímicas con el fin de optimizar la posología, mejorar la adherencia terapéutica, reducir el riesgo de aparición de reacciones adversas medicamentosas y minimizar las fluctuaciones plasmáticas. De esta manera, se opti- miza el tratamiento farmacológico de acuerdo con las necesi- dades de los pacientes y las situaciones clínicas particulares. En la segunda y tercera parte de este trabajo se hará foco en los grupos farmacológicos principales de la Neuropsicofarmacología que presentan diversos sistemas de liberación modificada.
Referencias bibliográficas
- 1. Brunton Lawrence. Goodman and Gil- man. Las bases farmacológicas de la terapéu- tica. Edición 13°. Año 2019. McGraw Hill Education.
- 2. Hernández Chávez Abel. Farmacología General. Una guía de estudio. Primera edi- ción. Año 2014. McGraw Hill Interamericana.
- 3. Isaza Carlos Alberto y colaboradores. Fundamentos de Farmacología en Terapéuti- ca. Sexta edición. Año 2014. Editorial Médi- ca CELSUS.
- 4. Viruete Cisneros Sergio. Manual de co- nocimientos básicos de farmacología. Univer- sidad de Guadalajara. Primera edición. Año 2015.
- 5. Rang HP, Dale MM, Ritter JM and Flower RJ. Rang y Dale Farmacología. Elsevier. 6° edición. Año 2008.
- 6. Pierre Mitchel Aristil Chéry. Manual de Farmacología Básica y Clínica. McGraw Hill. 5° edición. Año 2010.
- 7. Storpirtis S, Nella Gai M, Rossi de Cam- pos D, Goncalves JE. Farmacocinética básica y aplicada. Primera edición. Año 2011. Edi- tora Guanabara Koogan.
- 8. Ramírez Rigo María Verónica. Los siste- mas de liberación modificada de fármacos.
Consejo Nacional de Investigaciones Científi- cas y Técnicas (CONICET). Boletín electróni- co, número 9, año 2010.
- 9. Gómez Ayala Adela-Emilia. Manipulación de especialidades farmacéuticas. Farmacia profesional. 2007, 21(4): 44-48.
- 10. Berenguel Cook María del Rosario. Analgesia y vía sublingual. Revista Dolor, Clí- nica y Terapia. 2003, 2(6): 9-12.
- 11. Formas farmacéuticas de liberación modificada (FLM). Información Farmacotera- péutica de la Comarca. 2003, 11(8): 38-40.
- 12. Lastres García José Luis. Nuevos siste- mas orales de liberación modificada. Schiro- nia. 2002, 1: 63-71.
- 13. Moreno Frigols José Luis. Innovaciones farmacéuticas para la administración de me- dicamentos. Real Academia de Medicina de la Comunidad Valenciana. Valencia, España. Año 2002.
- 14. Especialidades farmacéuticas de libe- ración modificada y estereoisómeros. SACYL. Boletín de Información Terapéutica. 2004, 4: 1-4.
- 15. Formas farmacéuticas de liberación modificada y estereoisómeros. ¿Nos aportan algo en la práctica clínica? Boletín de In- formación Farmacoterapéutica de Navarra. 2005, 13(1): 1-9.
- 16. José María Suñe Negre. Nuevas aporta- ciones galénicas a las formas de administra- ción. Formación continuada para Farmacéuti- cos de Hospital. Páginas 28 a 69.
- 17. Paredero Domínguez JM. Nuevas for- mas farmacéuticas de liberación modificada: revisión y relevancia. SESCAM. 2007: 5-8.
- 18. Mazzoglio y Nabar M, Mejías Delamano A, Muñiz MM, Muñoz S, Magrath Guimet N. Psicofarmacología en esquemas para el equi- po interdisciplinario de salud mental. Segun- da edición. Año 2017. Editorial Impresiones Buenos Aires
- 19. Zarranz JJ. Neurofarmacología contem- poránea. Elsevier. Año 2011.
- 20. Stahl Stephen. Psicofarmacología Esencial de Stahl. Bases neurocientíficas y aplicaciones prácticas. Cambridge University Press. Cuarta Edición. 2013.
- 21. Zieher LM y colaboradores. Tratado de Psicofarmacología y Neurociencia: trastornos depresivos. Parte 2: farmacología y estrate- gias terapéuticas. Primera Edición, Buenos Aires. Editorial Sciens, 2010.
- 22. Concise Guide to Psychopharmacology. Lauren B Marangell, et al. Second Edition. 2006. American Psychiatric Publishing Inc.





No comments! Be the first commenter?