

La neurobiología de la esquizofrenia
Resumen
En este trabajo realizado sobre la base de revisiones bibliográficas, se destaca la función de los distintos neurotransmisores en el individuo normal y en el paciente esquizofrénico en las áreas claves de la corteza cerebral y de los ganglios de la base.
Las disfunciones de los neurotransmisores y sus interacciones fisio-patológicas ocasionadas por causas genéticas y epigené- ticas explicarían las diferentes hipótesis en el desarrollo de la neurobiología de la esquizofrenia.
Palabras clave
Esquizofrenia – Neurotransmisores – Disfunciones – Hipótesis – Genética – Epigenética – Neurobiología.
Pérez Vargas, María Sol. “Neurobiología de la esquizofrenia”. Psicofarmacología 2018;110:17-29.
Puede consultar otros artículos publicados por los autores en la revista Psicofarmacología en sciens.com.ar
Desde el punto de vista teórico, la esquizofrenia es una afección del neurodesarrollo en la que intervienen la genética y la epigenética.
Ha sido sugerido que un anormal desarrollo o plasticidad de la conectividad del hipocampo afectaría el desarrollo y la función de la corteza prefrontal. Meyer y Lindenberg y col. pu- sieron en evidencia una alteración en la conectividad funcio- nal en la esquizofrenia caracterizada por una hipofrontalidad y un incremento de la actividad témporohipocampal. Es decir, que un daño ambiental y/o una tardía expresión genética en la vida serían necesarias para desarrollar totalmente el síndrome esquizofrénico (1).
Durante la pubertad, en que se produce el proceso de “poda” o pruning, se jerarquiza a determinado grupo de neuronas en detrimento de un 30 a 40% que van a ser elimina- das. Este proceso dependerá de qué tan bien constituidas y mielinizadas estén las neuronas, ya que las mejor constituidas quedarán. Esto ocurre en un individuo normal (2).
En algunos casos, durante el período de la pubertad, antes de la etapa del pruning, es decir, en el período considerado prodrómico de la esquizofrenia, pueden observarse alteracio- nes de la conducta y trastornos motores como es la distonía de mano izquierda.
El proceso de pruning no se produce de la misma manera en un cerebro esquizofrénico que en un cerebro normal, por lo que pueden quedar neuronas anómalas, en lugar de neuronas bien constituidas. Esto se traduce en una menor cantidad de conexiones neuronales, debido a la menor cantidad de sinapsis y a que las neuronas son funcionalmente anómalas. Se encuentran alteraciones en los receptores y neurotransmisores, así como también presencia de espinas y dendritas aberran- tes. Por ello, la plasticidad neuronal se encuentra afectada.
Todas estas alteraciones ocurren en la CPFDL (corteza pre- frontal dorso lateral) particular y selectivamente (1).
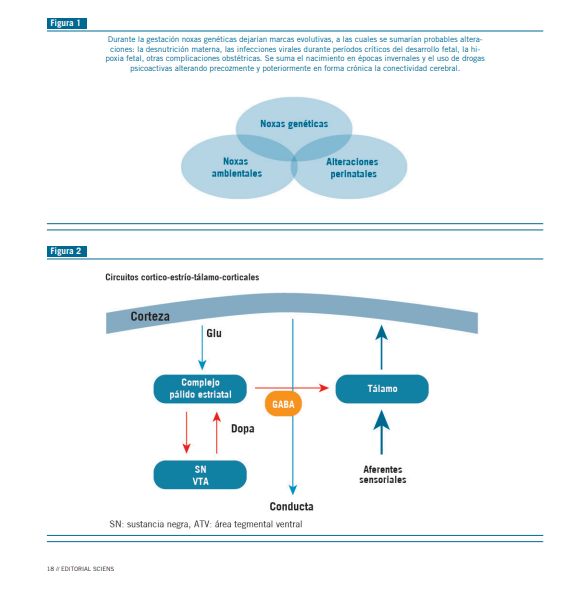
La conducta apropiada depende del equilibrio de los neuro- transmisores y del buen funcionamiento de receptores, a nivel del circuito córtico-estrio-tálamico-cortical.
A continuación se desarrollarán las hipótesis de la esquizofrenia actualizadas hasta la fecha:
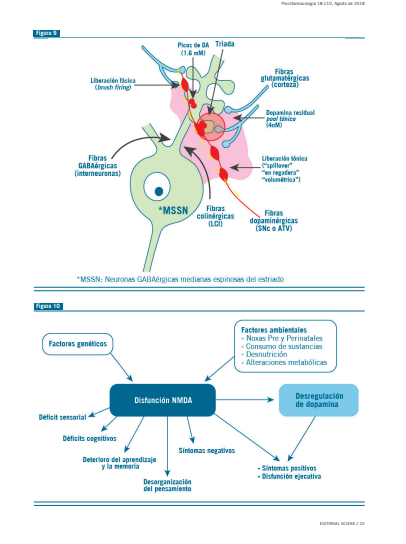
• Hipótesis de la hipofunción NMDA
El receptor NMDA necesita para actuar D-serina o glicina y que el Mg++ desbloquee el receptor y, de esta manera, pueda entrar Ca++ cuando se desencadena el potencial de acción.
Los canales NMDA son fundamentales en la generación de EPSP (potenciales post sinápticos excitatorios) en las in- terneuronas GABAérgicas candelabro y basket. La inhibición aguda de estos canales resulta en una menor inhibición GA- BAérgica sobre las células piramidales (Figura 3 y 4).
Este fenómeno de inhibición, activa a las neuronas glutamatérgicas con mayor síntesis y liberación de glutamato, que solo se vehiculiza post sinápticamente, vía AMPA/Kainato, y que su mayor aumento podría producir excitotoxicidad y convulsiones por la hipofunción NMDA.
Paralelamente a la hipofunción del receptor NMDA, que transmite a las interneuronas, que se sintetice menos GABA, hay un aumento de la síntesis y liberación de glutamato a nivel del
cortex prefrontal por la falta del freno GABAérgico (Figura 3). Las aberraciones de la señalización del receptor de glutamato NMDA fueron propuestas para ser consideradas como uno de los mecanismos subyacentes que contribuyen a los síntomas cognitivos de la esquizofrenia (3).
Los canales NMDA funcionan como “sensores” de la ac- tividad de las células piramidales y la ejercen a través de la GAD67 y la parvalbúmina de las interneuronas GABAérgicas. Si el sensor funciona mal, la interneurona sintetiza menos GABA y parvalbúmina en un intento de llevar la actividad de la célula piramidal a su nivel correcto, que en este contexto, resulta mal adaptativa, generando una sobreactividad de las células piramidales con aumento de la síntesis y liberación de glutamato, que determinaría el síndrome esquizofrénico (2) (Figura 3).
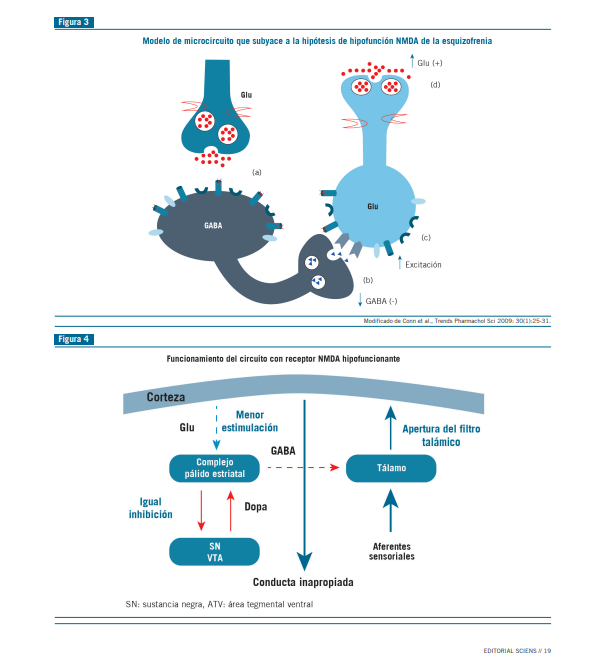
Habría menos Glu hacia el complejo pálido-estriatal y, por lo tanto, hay menos GABA hacia el tálamo. Esto determina que haya una mayor llegada de las aferencias sensoriales glutamatérgicas hacia la corteza, por falla del equilibro en el filtro talámico, determinada por la menor llegada de GABA, produciéndose una alteración en la conducta (Figura 4).
• Hipótesis dopaminérgica
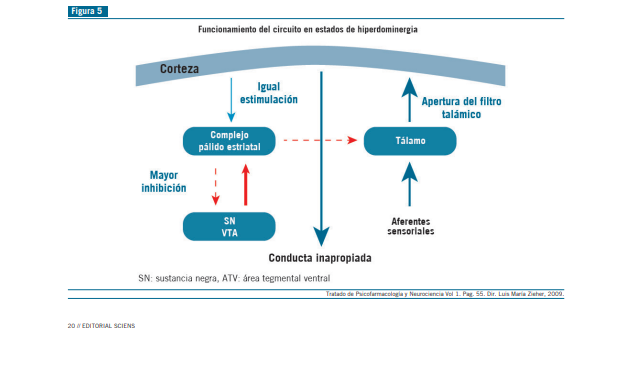
Respecto de la dopamina, (hipótesis de Carlsson en la década de 1960), Kapur formula la idea de las “saliencias aberrantes” de los eventos ambientales (delirios), o de repre- sentaciones internas (alucinaciones) otorgando un rol clave al estado hiperdopaminérgico desrregulado (2).
Existiría una hiperdopaminergia en la vía mesolímbica de los esquizofrénicos, responsable de las alucinaciones y delirios y una hipodopaminergia mesocortical responsable de la sinto- matología negativa y cognitiva, afectando en este último caso la working memory (habilidad para mantener transitoriamente en orden el material cognitivo, para guiarlo o conducirlo a una respuesta posterior) así como el alerta, vigilancia, aprendizaje verbal y no verbal (visual) y el razonamiento (Figura 6).
La hiperdopaminergia mesolímbica inhibe a las interneu- ronas, determinando que haya menos GABA hacia el tálamo, produciéndose un desequilibrio a favor de las aferencias sen- soriales glutamatérgicas, con apertura del filtro talámico, oca- sionado una conducta inapropiada.
En la CPF, el altamente organizado input DA sobre todas las dendritas de las neuronas piramidales, en los primates provee una manera de regular cercanamente la gran cantidad de afe- rentes glutamatérgicos que las neuronas piramidales reciben. La acción inhibitoria en individuos normales de la DA en la
CPF ocurriría por la excitación dopaminérgica de las interneu- ronas gabaérgicas que reciben inervación directa DA (4).
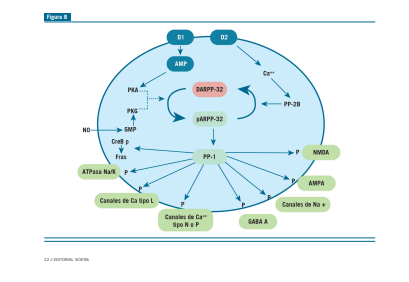
En el cuadro 6 tenemos integranción glutamato y dopamina en la CPF.Hay una disminución de la dopamina en CPF a través de la disminución de la activación del R D1 en la esquizofrenia (3). La activación del receptor D1 facilita la respuesta de NMDA por medio de Ca2+ y la proteína quinasa A y las interacciones sinérgicas entre los dos receptores son críticas para la excitabilidad de la neurona piramidal y la función de la CPF (3) (Figura 7). La DA, a través del receptor D1 y de la PK dependiente de AMPc, estimula la fosforilación de la DARPP-32 (Figura 8). La DARPP-32 cumple un rol central en la mediatización de la transducción de la señal en las neuronas medio espinosas del estriado y en la CPF. Ocuparía un lugar de “llave” regulatoria, jugando un rol importante en los cambios patofisiológicos de la función glutamatérgica y dopaminérgica de la CPF en la esquizofrenia.
En estado fosforilado, pero no en el desfoforilado, la DAR- PP-32 inhibe a la mayor serina/treonina protein fosfatasa 1 (PP1). A través de esta inhibición, la DARPP-32 controla el estado de fosforilación y de actividad fisiológica de distintas proteínas, canales de Na+ voltaje dependiente, canales de Ca++ voltaje dependiente tipo L, P y N, la bomba de Na+/K+ ATPasa dependiente, el receptor NMDA tipo NR1, el receptor AMPA, el receptor GABA A y los factores de transcripción CREB p-fras (4).
En el estriado es donde se encuentran las neuronas me- dioespinosas (Figura 9).
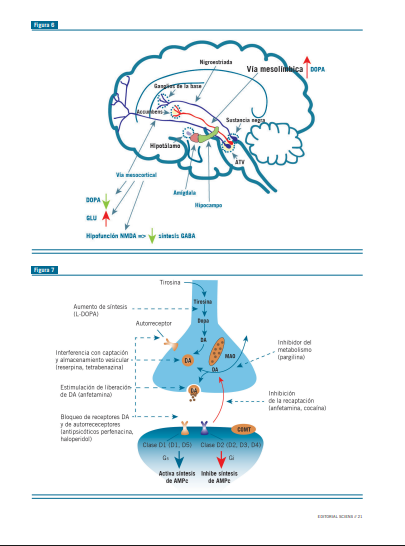
A nivel estriatal, en donde también actúa la DARP 32, se observa una tríada: las fibras glutamatérgicas provenientes de la corteza, las dopaminérgicas provenientes del ATV (área tegmental ventral) y las neuronas GABA, que son el principal output del estriado (Figura 9). La liberación tónica de Dopa se realiza desde las varicosidades grandes asinápticas. Las pe- queñas varicosidades, liberan la Dopa en forma fásica (burst firing), potenciales de acción rápidos y repetidos provenientes del ATV y de la SN (sustancia negra). La liberación fásica no ocurre en condiciones de reposo. La liberación tónica se halla bajo control glutamatérgico proveniente de la vía cortico-es- triatal. La DOPA fásica depende de la despolarización de los somas mesencefálicos. La liberación tónica sería la encargada de jerarquizar estímulos, ya que frena la liberación fásica de Dopa ante estímulos no novedosos (4).
Disfunción NMDA y desregulación de DOPA (Figura 11)
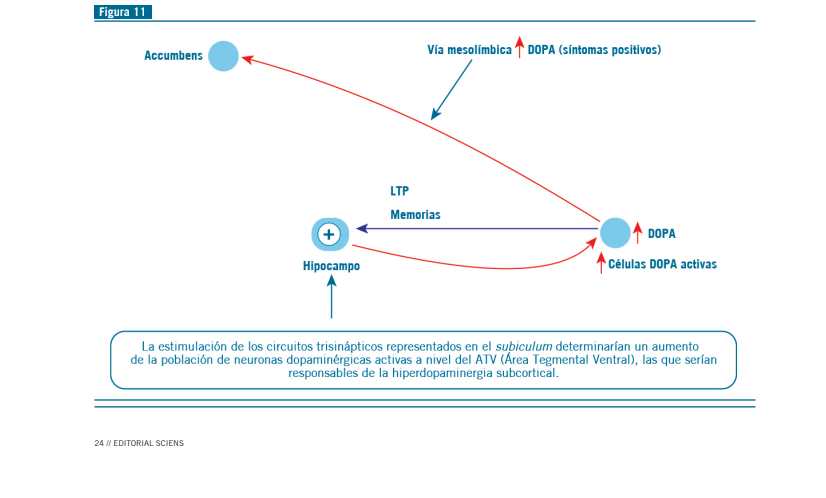
A esto se suma que la estimulación de los circuitos trisináp- ticos representados en el subiculum determinarían un aumen- to de la población de neuronas dopaminérgicas activas a nivel del ATV (área tegmental ventral), las que serían responsables de la hiperdopaminergia subcortical (esto se demuestra inac- tivando el subiculum), lo que indica que la región hipocampal es necesaria para producir el estado hiperdopaminérgico, en especial, la ideación delirante en psicosis esquizofrénicas y
no esquizofrénicas.
Las células del ATV proyectan al hipocampo e intervienen en la consolidación de la potenciación de largo plazo (LTP) y la entrada de información en memorias de largo plazo (1).
Por imágenes, se detecta mayor cantidad de dopamina en la corteza cingulada anterior, estriado y otras áreas del cere- bro, antes de desencadenarse la enfermedad, por lo que la indicación de antipsicóticos en la etapa previa prevendría la aparición de la enfermedad. No están indicados en la actua- lidad (5).
A su vez, en la CPF habría una disminución de DAT (la pro- teína transportadora de dopamina) y NET (la proteína trans- portadora de noradrenalina), a lo que se suma la actividad de la enzima COMT (catecol-O-metil transferasa) cuya isoforma Val-COMT (valina) tiene alelos susceptibles para esquizofre- nia. Variaciones en la actividad de la COMT, localizada en 22q11, tendría efectos específicos neurobiológicos en CPF.
Esta isoforma Val-COMT tiene una actividad enzimática del 75%, a diferencia de la isoforma Met-COMT (metionina) que tiene una actividad del 25%. De esta manera, se acelera el catabolismo de la dopamina por la forma VAL- COMT, sumado a la disminución de la recaptación de dopamina por el DAT, por lo que habría menos dopamina en la CPF, lo que expli- caría los síntomas negativos y cognitivos, ya que la presencia de la dopamina actúa en la cognición: memoria de trabajo y procesamiento de la información (2).
- Hipótesis gabaérgica
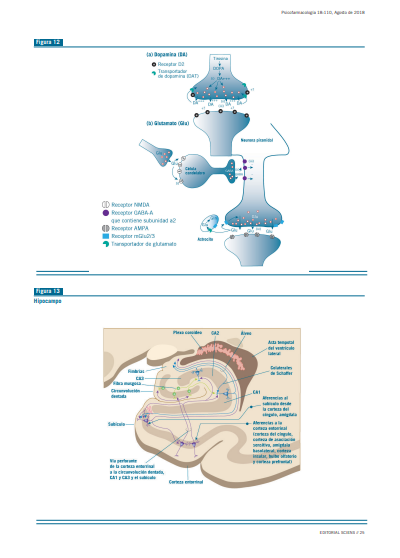
Las alteraciones no ocurren en todos los tipos de interneu- ronas GABAérgicas corticales (que son de seis clases diferen- tes), sino que se reducen a las interneuronas de tipo cesto (basket) y candelabro (chandelier) (Figura 12). Ambos tipos de neuronas contienen una proteína fijadora de calcio (Ca2+), la parvalbúmina y hacen sinapsis en la región perisomática de las células piramidales.
Estas neuronas disparan en altas frecuencias (fast-spiking) y hacen blanco en el segmento inicial del axón (IAS) de las neuronas piramidales, por lo que cumplen un rol clave en el control de las propiedades de disparo de las redes cerebrales y en particular, en las oscilaciones gama (30-100 Hz). Estas se detectan en el EEG reflejando la descarga sincronizada de grupos de células piramidales. En la esquizofrenia hay una reducción de oscilaciones gamma, la cual está correlacionada con la severidad de los síntomas negativos.
Si bien existe una modesta reducción en el número de interneuronas, los cambios mayores ocurren en la concentración de proteínas particulares, como la GAD67 y la parvalbúmina. Estos cambios son notorios en muchas regiones corticales y en el hipocampo, sobre todo en las regiones CA2/3 y en el striatum oriens de la región CA1 (Figura 13). Por otra parte, habría además un hiperflujo en el hipocampo, en la región CA1 en la esquizofrenia.
• Hipótesis serotonérgica
Con respecto a la acción de la serotonina, en la esquizofre- nia habría una interacción molecular entre el receptor 5HT2 y el mGluR2. Ambos receptores están acoplados a proteínas
G diferentes, y forman complejos funcionalmente relevantes. Los agonistas de receptores mGluR2 incrementan la afinidad de los alucinógenos por su sitio de fijación 5HT2A. Los alucinógenos LSD y psilocibina tienen similitud estructural con la serotonina. La activación de los mGluR2 suprime los efectos de los alucinógenos sobre la señalización celular. Además, los antagonistas NMDA como la fenilciclidina o la ketamina reproducen la signo-sintomatología de la esquizofrenia.
En pacientes esquizofrénicos hay niveles aumentados de 5HT2A y disminución de mGluR2, lo que predispone al es- quizofrénico a un patrón alucinógeno de señalización.
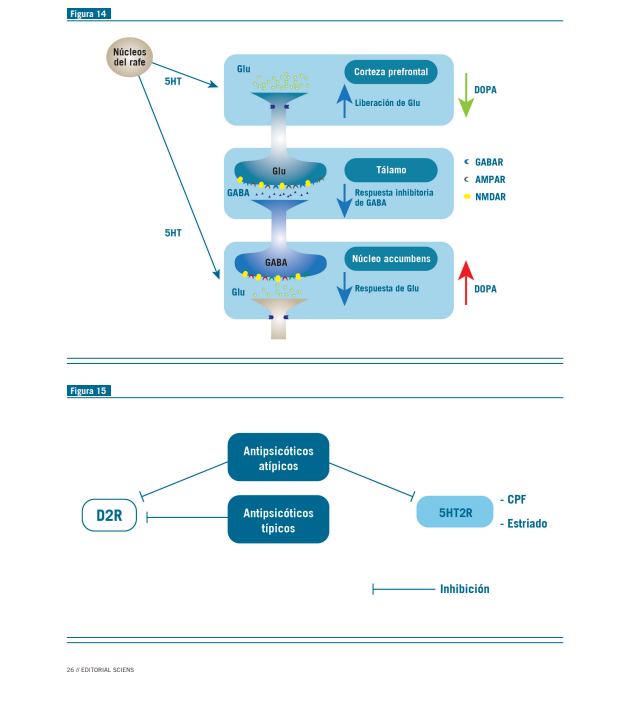
En pacientes añosos habría menor densidad de los componentes 2AR-mGluR2 y esto guardaría relación con la disminución de alucinaciones y delirios, que se observa en ancianos esquizofrénicos. Estos hallazgos tienden a unificar la hipótesis glutamatérgica y serotonérgica (6). Fisiológicamente, los receptores serotonérgicos modulan la liberación de DA en el estriado ventral, sin modificar la disponibilidad de DA en el estriado dorsal (Figura 14). Se libera 5HT (serotonina) desde los núcleos del rafé que estimulan la liberación de glutamato y este, a su vez, libera dopamina en la vía mesolímbica produciendo la sintomatología psicótica en la esquizofrenia. El receptor 5HT2A predomina en la CPF y en esta corteza la dopamina regula al Glu (Figura 16). Los bloqueantes de los receptores 5 HT2 inhiben la liberación de Glu desde terminales glutamatérgicas en el estriado y en la CPF, por lo que por dicha vía también modulan la neurotransmisión DA (Figura 15).
Por esta razón es el beneficio del bloqueo de los R 5HT2A por los antipsicóticos atípicos, ya que disminuyen, al internalizar el receptor, la población de estos en las espinas dendríticas de las células piramidales (7) (Figura 15).
• Hipótesis colinérgica
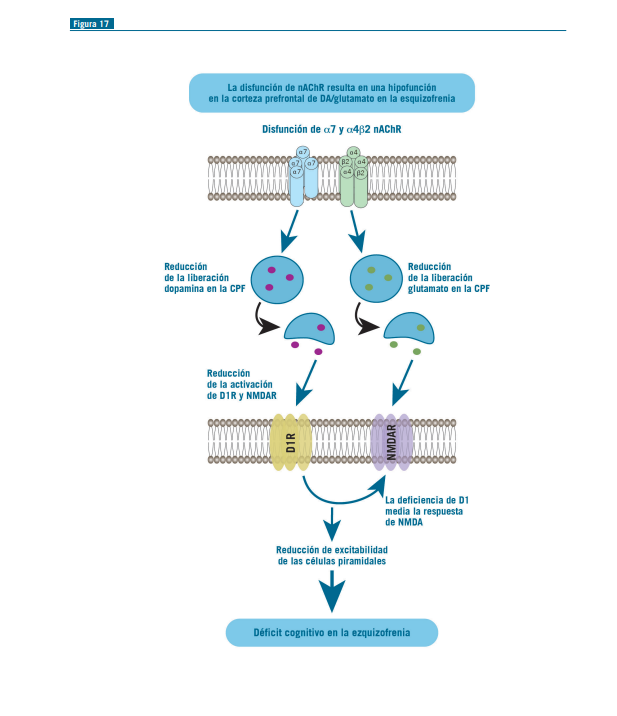
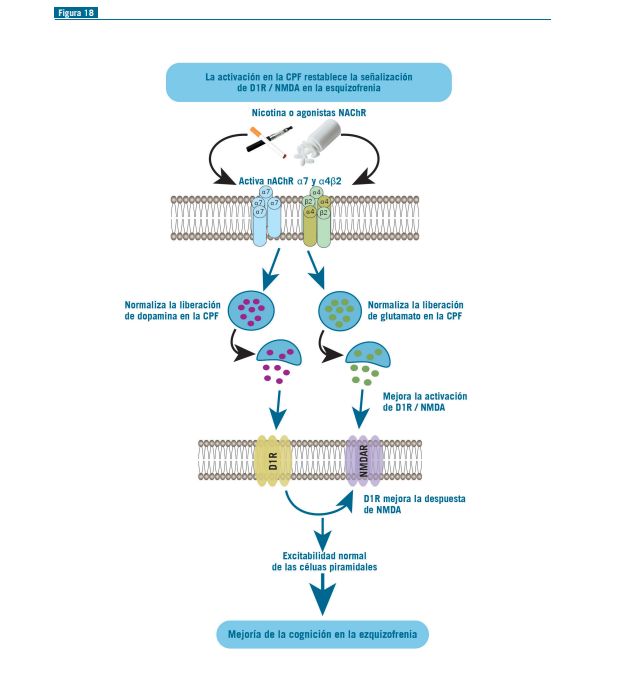
Los receptores nicotínicos de tipo a7 (pentámero homomérico) se concentran en las interneuronas, pueden despolarizarlas y aumentar la liberación de GABA (Figuras 17 y 18).






No comments! Be the first commenter?