

Modulación del ARN en la farmacología cardiovascular Parte 1: farmacología básica
Resumen
A medida que han avanzado el conocimiento del código genético humano, se han intentado desarrollar fármacos que lo modulen. En esta serie de dos artículos, se revisan la farmacología básica y aplicada a las enfermedades cardiovasculares de los fármacos recientemente descubiertos que tienen como sitio diana al ARN.
Palabras clave
Enfermedad cardiovascular – Ácido ribonucleico – Oligonucleótidos antisentido – ARN pequeño de interferencia – Terapia génica – N-acetilgalac- tosamina.
Introducción
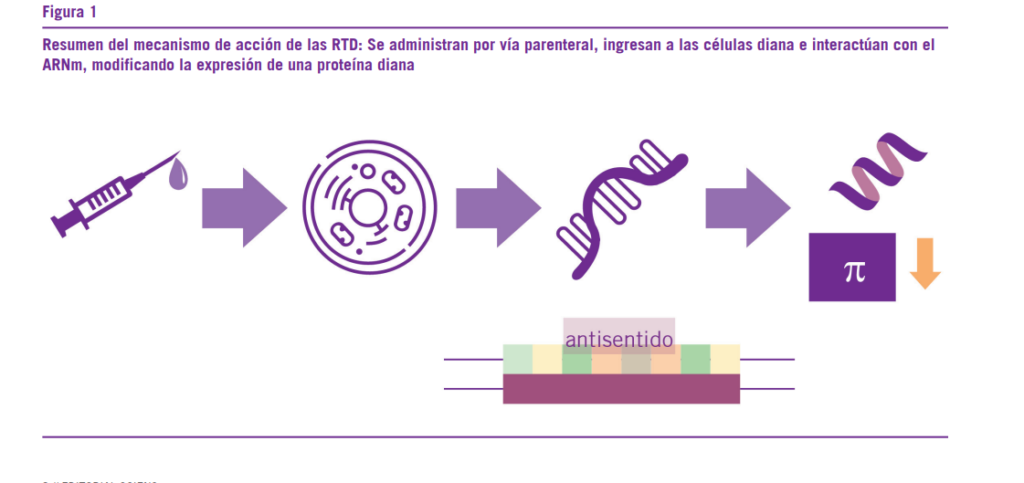
Las drogas cuyo blanco es el ARN (RNA targeted drugs, RTDs por sus siglas en inglés) son un tipo de terapia génica que permite el diseño de fármacos que pueden alterar la expresión de proteínas celulares, útiles para el tratamiento de una gran cantidad de enfermedades. Si bien los primeros desarrollos datan desde hace más de 20 años, es en los últimos tiempos en donde han tenido un resurgimiento en su relevancia, con fármacos como el Mipomersen y el Nusinersen. En el ámbito de la farmacología cardiovascular, se encuentran disponibles o en desarrollo más de una decena de RTDs, para el tratamiento de diversas patologías desde la Hipercolestero- lemia hasta la amiloidosis cardíaca.
De una forma similar a los anticuerpos monoclonales, las RTDs comparten un mecanismo de acción general, con di- ferencias en los efectos farmacológicos según la diana a la cual estén dirigidos. Estas drogas interactúan con el ADN o el ARNm celular y modifican la síntesis de proteínas. Dentro de esta familia de fármacos, encontramos dos subgrupos im- portantes: los Oligonucleótidos Antisentido (ASO, antisense oligonucleotides) y los ARN pequeños de interferencia (siRNA small interfering RNA).
A continuación se describirán los aspectos farmacológicos generales.
Sterba JJ, Song S, Piccinato A, Robino ON, Ramos MC, Muslera C, Zaidel EJ. “Modulación del ARN en la farmacología cardiovascular. Parte 1: farmacología básica”.
Farmacología Cardiovascular 2023;58:4-9 .
farmacología cardiovascular 58 | septiembre de 2023
Estructura y Mecanismo de Acción de los ASOs/siRNA
En líneas generales, las RTDs se valen de la interacción en- tre cadenas sintéticas de oligonucleótidos y cadenas de ARN celulares, para generar sus acciones. Estas drogas pueden ser divididas en dos grupos según su estructura, por un lado, los oligonucleótidos antisentido (ASOs, Antisense Oligonucleoti- des), que son cadenas de una sola hebra de oligonucleótidos que se unen a hebras de ARN complementarias a su secuen- cia, y por otro lado los ARN de interferencia pequeños (siRNA, small interfering RNA), cadena de dos hebras conformada por oligonucleótidos que utilizan la enzima AGO2.
Ambos grupos tienen como fin alterar el fenotipo de la en- fermedad mediante la degradación selectiva de un ARN dia- na, llevando a la disminución de la síntesis de proteínas im- plicadas en ciertas patologías.
Partiendo de una proteína que participe en una determi- nada enfermedad, se busca la secuencia de ARNm que la codifica, luego se sintetiza en el laboratorio una secuencia complementaria (antisentido) a la misma, la cual es adminis- trada e ingresa a las células, donde se une a su ARNm. Este ARN no puede ser traducido hacia una proteína y así se logra interferir en el curso de la enfermedad.
Sentido y antisentido hacen referencia a la polaridad de una cadena de nucleótidos. La cadena sentido (codificante o positiva) es aquella que tiene la secuencia de nucleótidos que, al ser leída por los ribosomas, codifica para una proteína. Por otro lado, la cadena antisentido presenta una secuencia de nucleótidos contraria (no codificante o negativa) a la ca- dena codificante. Por apareamiento de bases, ambas cade- nas pueden unirse formando una doble hebra. Tanto los ASOs como los siRNA están formados por cadenas antisentido que de una forma u otra interactúan con las hebras codificantes del material genético de la célula. Otro concepto importante es el de oligonucleótido, el cual hace referencia a una cadena de nucleótidos menor a las 50 bases nitrogenadas de largo. Tanto los oligonucleótidos antisentido como los ARN de in- terferencia pequeños son, en esencia, oligonucleótidos. En la tabla 1 se describen las características de estas moléculas.
Los ASOs se encuentran constituidos por cadenas de nu- cleótidos simples (de una sola hebra) unidos mediante en- laces fosfodiéster que forman una columna o backbone. La longitud aproximada de estas cadenas es de entre 15 y 20 nucleótidos. Por otro lado, los siRNA están formados por dos cadenas de nucleótidos: una cadena antisentido “guía” y una hebra “pasajera” que actúa como un transportador de la an- terior. Tienen una longitud de entre 19 y 22 pares de bases y un peso mayor a los ASOs.
La estructura de estos fármacos tiene implicancias impor-
tantes no sólo para su mecanismo de acción, sino también para su farmacocinética y efectos adversos. Por ejemplo, las cadenas más largas corren el riesgo de ser detectadas por el sistema inmunológico y disparar respuestas celulares como si de un virus se tratase. La modificación de los grupos fosfato (PO), los azúcares y la conjugación con monosacáridos (ver más adelante) son estrategias muy utilizadas para modificar la farmacodinamia y farmacocinética de estas drogas.
Según su estructura, los diferentes fármacos antisentido se clasifican en generaciones. La primera generación se caracte- riza por la modificación en los grupos fosfato de la columna. La segunda generación abarca los fármacos que tienen grupos al- quilo en sus nucleótidos. La tercera generación se compone de los oligonucleótidos más estables y más efectivos (in vitro), se caracterizan por presentar otras modificaciones químicas que mejoran sus propiedades farmacodinámicas principalmente.
Hablando específicamente de los mecanismos de acción que pueden ser utilizados por los RTDs, encontramos dos caminos principales: Degradación mediada por ocupación y mecanismos sólo de ocupación. En la degradación mediada por ocupación, luego de la unión de los RTDs al ARN diana se produce una degradación enzimática del ARN. Este me- canismo es utilizado tanto por los ASOs como por los siRNA. Las principales enzimas implicadas son la Ribonucleasa H1 (principalmente para los ASOs, y Argonaute 2 (principalmente para los siRNA). La enzima RNasa H1 es una endonucleasa (corta los enlaces interiores de la cadena de ácidos nucleicos) que pertenece a la familia de las ribonucleasas H, su función en las células es regular la expresión de distintos ARNm to- mando como molde secuencias de ADN. En el caso de los ASOs, el mecanismo de acción implica al siguiente proceso: El ASO ingresa a la célula y se encuentra con la RNasa H1, la enzima reconoce una secuencia de nucleótidos en el ASO y forman un complejo enzima-RTD, que localiza el ARN al cual está dirigida la terapia, la enzima reconoce la secuencia com- plementaria en el ARN mensajero, el cual es clivado en dife- rentes sectores por la RNasa H1 y se degrada, y finalmente la proteína codificada por el ARN mensajero no es sintetizada.
Argonaute 2 es una endonucleasa que reconoce duplas de ARN-ARN, y que se encuentra implicada en la respuesta ce- lular contra los virus. Es una enzima que permite pocos cam- bios químicos en las cadenas del siRNA, lo que significa que su estructura tiene menos posibilidades de ser modificada. En el caso de los siRNA, el mecanismo de acción implica el siguiente proceso: El siRNA ingresa al citoplasma y es recono- cido por Ago2, que procede a separar las hebras y permitir la degradación de la cadena “pasajera”. Luego, Ago2 forma un complejo llamado RISC con la cadena “guía”, que detecta el ARNm complementario a la cadena “guía” del siRNA. Ago2 procede a hidrolizar la cadena blanco de ARN, y el complejo RISC permanecerá activo por un tiempo prolongado, degra- dando más cadenas diana.
En los mecanismos solo de ocupación, la unión del RTD al ARN diana impide su traducción sin utilizar enzimas, gracias a lo que se conoce como bloqueo estérico en donde el RTD impide físicamente la acción de los ribosomas. Solamente los ASOs utilizan este mecanismo. El principal resultado del blo- queo estérico consiste en la detención de la traducción o la alteración de estructuras importantes dentro del ARNm que impiden el inicio de la traducción. De esta forma, al no ser traducido el ARNm diana, disminuye la expresión de la pro- teína que codifican.
Un dato importante es que también se pueden diseñar ASOs capaces de aumentar la síntesis de una proteína. Estos ASOs están dirigidos contra los microARN (mi-ARN), pequeñas se- cuencias de nucleótidos que la célula utiliza para degradar al ARNm y evitar su transcripción. Si se diseña un ASO capaz de bloquear a los mi-ARN, se logrará evitar su efecto repre- sor, aumentando así la cantidad de proteínas que se traducen (up-regulation). Estaríamos “inhibiendo” a un “inhibidor”.
Farmacocinética
En líneas generales, tanto los siRNA como los ASOs pre- sentan una muy baja biodisponibilidad oral, no penetran la barrera hematoencefálica (BHE), tienen unión a proteínas plasmáticas, utilizan mecanismos de ingreso a las células, se internalizan en endosomas y carecen de metabolismo por ci- tocromos. La administración de estas drogas local se ve deter- minada por la patología para la cual fue diseñado el fármaco, siendo relevantes para las patologías sistémicas la utilización de las vías endovenosa (EV) y subcutánea (SC). Ambas pre- sentan una buena biodisponibilidad (BD) pero la presencia de nucleasas en la piel puede disminuir levemente el porcentaje de fármaco absorbido en la vía subcutánea.
Un inconveniente fundamental en lo que respecta a las RTDs es garantizar las concentraciones necesarias en las cé- lulas que expresan el ARN diana. Para mejorar este aspecto y permitir la llegada hacia órganos específicos, se utiliza la conjugación con distintas moléculas. Una de las más impor-
tantes es la N-acetilgalactosamina cuyo receptor se expresa de forma exclusiva en los hepatocitos. Abreviado como Gal- Nac, este monosacárido se conjuga con los RTDs y permite su interacción con los receptores ASGPR-3 que se encuentran expresados en la superficie de estas células lo que determina su rápida endocitosis. Así, se logra mejorar de gran manera el perfil farmacocinético de estos fármacos. Según estudios, la conjugación con GalNac sería la responsable de una marcada disminución de la cantidad y gravedad de efectos adversos en comparación con RTDs no conjugados por su exposición sis- témica. Se ha demostrado que, en drogas que tengan acción a nivel hepático, la conjugación con GalNac aumentaba hasta 30 veces la magnitud de la respuesta observada en los parti- cipantes a mismas dosis. Estos hallazgos permitieron elaborar esquemas de tratamiento con dosis considerablemente más bajas, lo que finalmente disminuye la exposición sistémica y por ende los efectos adversos, sin disminuir la efectividad.
Los ASOs presentan en su estructura regiones hidrofílicas correspondientes a los grupos PS (fosforotioato) e hidrofóbi- cas, correspondientes a las bases nitrogenadas. Es por ello que se comportan como moléculas anfipáticas. En lo que respecta a su administración, su estructura permite generar drogas que se administren por casi todas las vías: subcutánea, endovenosa, oral e inhalatoria. De todas formas, la adminis- tración oral cuenta con una baja biodisponibilidad y no parece ser una alternativa útil.
En ASOs que presenten grupos PS y una longitud mayor a 12 nucleótidos la distribución sigue un patrón similar: presen- tan unión a proteínas plasmáticas con una distribución am- plia, acumulación en tejidos ricos en lípidos (hígado, riñón, tejido adiposo, médula ósea y bazo) y una vida media plas- mática de 1-2 horas. La vida tisular ronda las 3-4 semanas y la administración en dosis altas permite saturar los depósitos, alcanzando semividas de hasta 6 meses en los tejidos. La con- jugación con GalNac permite el envío preferencial al hepatoci- to, lugar casi exclusivo de expresión de su receptor, ASGPR-3. Al interactuar con su ligando, el receptor de GalNac permite la endocitosis del ASO. Esta modificación incrementa hasta 30 veces el efecto de la droga en comparación a las mismas
Tabla 1
Diferencias entre los dos grandes grupos de RTDs
| Característica | Oligonucleótidos antisentido | ARN de interferencia pequeños |
| Estructura de cadena | Cadena simple: antisentido | Cadena doble: sentido y antisentido |
| Longitud y peso | 15-20 pb, ± 7 KDa | 19-22 pb, ± 14 KDa |
| Monómeros utilizados | Ribonucleótidos y desoxirribonucleótidos | Ribonucleótidos |
| Mecanismo de acción | Degradación mediada por ocupación bloqueo estérico | Degradación mediada por ocupación |
farmacología cardiovascular 58 | septiembre de 2023
dosis sin conjugar. Siguiendo los pasos de la conjugación con GalNac, se están estudiando otras alternativas para el envío dirigido de estos fármacos.
El ingreso en las células está mediado por varios mecanismos de endocitosis como aquellos mediados por clatrina, caveolina y micropinocitosis. Una vez ingresan a la célula, se localizan en endosomas tempranos. Luego de la maduración hacia endo- somas tardíos, los ASOs son liberados hacia el citoplasma o el núcleo para ejercer su efecto. El ingreso de estos fármacos al sistema nervioso central (SNC) es nulo, ya que su estructura no permite cruzar la barrera hematoencefálica (BHE).
La vida media tisular de los ASOs suele determinar la dura- ción de acción de estos, eso explica la relevancia de los me- canismos que degradan a estas drogas. La hidrólisis mediante endonucleasas consiste en el metabolismo de estas sustan- cias como primer paso. Los fragmentos resultantes, carentes de función y más hidrosolubles, pueden ser degradados por exonucleasas o en algunos casos eliminados de la célula y excretados en orina. También en la orina pueden detectarse ASOs intactos. No parece ser necesario el ajuste de dosis en pacientes con disminución de la función renal. Al no presen- tar metabolismo mediante el citocromo P450, limita en gran medida las interacciones farmacológicas con otras drogas.
Los siRNA, a diferencia de los ASOs, poseen una estructura de doble cadena que permite la interacción entre sus regiones hidrofóbicas, exponiendo solamente las porciones hidrofíli- cas. A priori esto limita su administración a la vía endovenosa, previa formulación con lípidos catiónicos (como los presen- tes en vacunas de ARNm contra COVID-19). Sin embargo, la conjugación con GalNac permite su administración por vía subcutánea y reduce los efectos adversos relacionados con la inflamación generada por los lípidos catiónicos. El ingreso a las células sigue pasos similares a los ASOs. Estos fármacos tampoco ingresan a SNC. Los siRNA no conjugados ni formu- lados con lípidos tienden a ser rápidamente filtrados en el riñón y excretados por orina de forma inalterada. No parece ser necesario ajustar la dosis en pacientes con disminución de la función renal.
El metabolismo de los siRNA es compartido con el de los ASOs y su vida media tisular es de aproximadamente 3 sema- nas, pudiendo utilizar el mismo principio de saturación para alargarla. La duración de acción también es similar a la vida media tisular, aunque también puede variar según el tiempo que permanezca Ago2 cargada con el siRNA. Más allá de lo expuesto, algunos trabajos destacan que la duración de ac- ción de un ASO y un siRNA dirigidos a la misma diana de ARN, es igual.
Efectos adversos e interacciones
El conocimiento respecto a sus efectos adversos (EAs) e interacciones se encuentra en continuo crecimiento. La ma- yor parte de los EA descritos parecen tener un componente inmunomediado y no depender del mecanismo de acción de las drogas, sino de su estructura química. No debemos olvidar las reacciones adversas locales en el sitio de inyección de formulaciones subcutáneas. Los ASOs que más se asocian a los efectos adversos locales son los que contienen grupos PS. Ellos también pueden presentar un incremento en la activa- ción y agregación plaquetaria por interactuar con la glicopro- teína IV. Además, la unión con otras proteínas puede llevar a la inhibición de la vía intrínseca de la coagulación. Por otro lado, los grupos PS pueden activar la vía alterna del comple- mento, con la consecuente cascada inflamatoria asociada.
Esta cascada inflamatoria es responsable de la aparición de glomerulonefritis y vasculitis, estudiados en modelos anima- les. En el caso de seres humanos, se ha demostrado que la presencia de enfermedad renal predispone para el desarrollo de glomerulonefritis posterior a la administración de ASOs. Sin embargo, en personas sanas, la aparición de glomeru- lonefritis es muy poco frecuente. Se ha propuesto que las concentraciones plasmáticas tienen un umbral para el desa- rrollo de este efecto adverso por lo que diseñar un protocolo de administración que no supere este umbral debería reducir el riesgo. La posibilidad de conjugar con GalNac a las RTDs destinadas al hígado parece ser una buena alternativa, ya que permite limitar la concentración máxima plasmática (Cmax) y
Tabla 2
Diferencias entre los dos grandes mecanismos de acción de los RTDs
| Característica | Degradación mediada por ocupación | Mecanismos sólo de ocupación |
| Fundamento | Degradación enzimática (RNasa-H1, Ago2) | Bloqueo estérico, no requiere enzimas |
| Efecto farmacológico | Disminución de la expresión de una proteína | Disminución o aumento de la expresión de una proteína |
| Grupos farmacológicos | Oligonucleótidos Antisentido, ARN de interfe- rencia pequeños | Oligonucleótidos Antisentido |
aumentar la concentración de droga en los hepatocitos.
Las modificaciones químicas que abarcan a la ribosa (se- gunda generación) pueden mejorar o empeorar los perfiles farmacocinéticos y farmacodinámicos evitando los efectos pro-inflamatorios característicos de las RTDs que poseen gru- pos PS. Sin embargo, en varios trabajos que utilizaron una modificación llamada 2’-MOE se ha evidenciado la aparición de trombocitopenia en dos variantes que podrían tener una fisiopatogenia distinta.
La primera forma de trombocitopenia se ha descrito como leve, reversible y dosis dependiente. Parece ser más frecuen- te en las RTDs de primera generación que poseían tanto PS como 2’-MOE. Esta forma no parece tener repercusión clínica en lo que respecta a sangrados. La segunda forma de trombo- citopenia se cataloga como severa (en monos la gravedad de las hemorragias requirió suspensión del fármaco) y es menos frecuente que la primera. Se observa en caso de administra- ción repetida, y si bien el recuento plaquetario se eleva cuan- do se suspende el fármaco, su re-administración produce una recaída. Resta definir si es una reacción adversa de tipo dosis independiente como sugieren algunos estudios con bajo nú- mero de animales involucrados. En un estudio, se encontraron anticuerpos anti-plaquetarios en un bajo número de pacientes.
A la fecha, no se ha podido establecer con certeza la habili- dad de predecir el desarrollo de trombocitopenia mediante los estudios de farmacología pre-clínica. Se ha documentado en un estudio el fallecimiento de un participante por hemorragia intracraneal producto de una trombocitopenia grave, y en otro estudio con pacientes pediátricos que presentaban tumores cerebrales, la muerte de dos participantes por sangrado del tumor luego de trombocitopenia. Otras modificaciones estruc- turales parecen estar asociadas al desarrollo de efectos adver- sos a nivel renal y hepático.
La trombocitopenia severa parece responder a varios me- canismos sin uno que claramente predomine sobre el resto. Dentro de las hipótesis que se tienen en cuenta para explicar el desarrollo de trombocitopenia, los datos disponibles su- gieren que la secuencia codificada en el ASO podría tener relevancia. También podría estar implicada la unión a las pla- quetas circulantes, dada la carga negativa de estas drogas, de forma similar a la trombocitopenia inducida por heparina. Otras opciones que se tienen en cuenta es la presencia de co- morbilidades que incrementan el riesgo y el hiperesplenismo producto de la activación del sistema inmune. A la fecha no se ha documentado la aparición de trombocitopenia severa causada por la administración de siRNAs, aunque esto puede deberse a la reciente inclusión en estudios de farmacología clínica. De todas formas, los componentes lipídicos en los cuales se formulan los siRNA pueden causar trombocitope- nias inmunomediada. La trombocitopenia se ha documentado con varios ASOs.
En lo que respecta a los siRNA, reportes de estudios de Fase II-III con Revusiran (para el tratamiento de la amiloidosis por transtiretina) han evidenciado, además de un aumento en la mortalidad del grupo con principio activo, aparición de neu- ropatía periférica. Se desconoce actualmente si hay relación entre ambos hechos y cuál es la fisiopatología de este tipo de efecto adverso (recordar como posible confundidor que en la amiloidosis por transtiretina es común el desarrollo de neu- ropatía). Una vez más, el sistema de envío por conjugación con GalNac podría dar lugar a una reducción en estos efectos adversos ya que permite llegar a niveles plasmáticos 30 veces menores con la misma eficacia.
Los efectos por dianas fuera de rango (off target effects) son aquellos producidos por la unión a dianas distintas a las que se buscan para el fármaco. A la fecha, no se han reportado reac-
ciones adversas en donde con certeza se haya documentado la participación de este mecanismo, pero creemos importante no dejar de mencionarlo por sus posibles implicancias en un futuro. La RNasa H1, utilizada por los ASOs, es una enzima muy específica para reconocer pares de ARN/ADN y clivar el ARN. Es muy poco frecuente que esta enzima logre unirse y clivar secuencias de ARN distintas a las complementarias a la secuencia de ADN.
En lo que respecta a Ago2, encontramos que esta enzima presenta una especificidad levemente menor en cuanto a sus sustratos ya que solamente utiliza una secuencia de 7 nucleótidos (la cual en teoría puede encontrarse en secuencias de ARNm de proteínas distintas). Otra posible fuente de efectos adversos se relaciona con las funciones de Ago2, ya que ésta se encuentra usualmente en actividad regulando la expresión de distintos ARN,por lo que su utilización por los siRNA podría alterar esta regulación. En ambas familias de RTDs se ha
descrito la citotoxicidad mediada por formación de complejos proteicos tóxicos entre las drogas y blancos intracelulares, resta definir su relevancia en la clínica. Dadas sus propiedades farmacológicas, las interacciones con otros fármacos son prácticamente nulas.
Conclusiones
Las nuevas terapias dirigidas al ARN se encuentran en pleno auge, y las ventajas que ofrecen son numerosas: Son fármacos con excelentes resultados particularmente en pacientes cuyas patologías no tenían anteriormente un tratamiento efectivo, con nulas interacciones, y con una larga duración de acción. Resta profundizar el conocimiento de los efectos adversos de estos fármacos, como de cualquier fármaco que se encuentra recientemente aprobado. La administración parenteral y los elevados costos iniciales son algunas de las barreras comunes a ellos que deberán considerarse. A pesar de ello, podemos afirmar que los RTDs son una familia de fármacos que ha llegado para quedarse, y sus resultados son prometedores, particularmente en el ámbito de la patología cardiometabólica.





No comments! Be the first commenter?