Revisión de su farmacología básica y clínica en el contexto del metabolismo lipídico. Parte 1
Las dislipemias, independientemente del tipo de lípido involucrado, son aterogénicas y contribuyen como factor de riesgo independiente a la aparición de la enfermedad cardiovascular isquémica. La introducción de las estatinas cambió radicalmente el tratamiento de las hipercolesterolemias. Si bien la mayoría de los pacientes responden a la terapia con estatinas, existen formas familiares sumamente resistentes y hay situaciones donde las estatinas no son bien toleradas o directamente están contraindicadas. El ácido bempedoico es un el primero de los inhibidores de la ATP citrato liasa (ACLy), enzima principal de la lanzadera del citrato la vía que devuelve sustratos al citoplasma desde la mitocondria para procesos anabólicos. Por ello, este fármaco podría resultar sumamente útil en las dislipemias pues corta el aporte de acetil-CoA para la síntesis de ácidos grasos y de colesterol hepática, precursores de lipoproteínas aterogénicas. Este articulo tiene como objetivo mostrar la farmacología básica y clínica del ácido bempedoico resaltando su seguridad y eficacia en la terapia de las dislipemias.
Autor
Dr. Héctor Alejandro Serra. Prof. Adjunto de Farmacología, Facultad de Medicina, UBA
Introducción
Dentro de las enfermedades no transmisibles, las de origen cardiovascular lideran en Argentina y en el mundo las causas de mortalidad (1) pues comparten factores de riesgo enmarcados en un estilo de vida común a las grandes comunidades tales como dietas hipercalóricas o con exceso de sodio, sedentarismo, tabaquismo, falta de descanso, estresores sociales y consumo problemático de sustancias. Estos factores conducen a nuevas situaciones con mayor riesgo y severidad como el sobrepeso, la obesidad y el síndrome metabólico que, si se sostienen, se retroalimentan avanzando hacia la disfuncionalidad y el daño orgánico manifiesto responsables de la morbimortalidad cardiovascular.
La hipertensión, las dislipemias y la hiperglucemia como manifestaciones del síndrome metabólico (2-5) subyacen asintomáticamente en la mayoría de los enfermos, por lo que al no ser advertidas sin una pesquisa sistemática y tratadas convenientemente, son las que, con el discurrir del tiempo, producen dicha disfuncionalidad.
Las dislipemias en más o hiperlipemias son el principal elemento desencadenante de la enfermedad cardiovascular isquémica por arteriosclerosis (6). Las hiperlipemias resultan en el aumento de los lípidos circulantes, colesterol (hipercolesterolemia), triglicéridos (hipertrigliceridemia) o ambos (formas mixtas) por encima de ciertos valores preestablecidos epidemiológicamente (6-8) y responden a causas genéticas (hiperlipemias primarias o familiares) o a enfermedades de base como diabetes, alcoholismo, hábitos alimenticios inapropiados o consumo de fármacos (hiperlipemias secundarias).
Las metas de todo tratamiento de las hiperlipemias consisten en reducir por medios farmacológicos y no farmacológicos los lípidos séricos por debajo de valores de corte dados (7), valores un poco más laxos para prevención primaria (lo que significa prevenir la aparición de un primer episodio isquémico cardiovascular como infarto agudo de miocardio, o accidente cerebro vascular y sus secuelas) que para prevención secundaria (lo que implica la necesidad de que no se produzca un nuevo episodio después de haber padecido un primer cuadro isquémico).
Por consiguiente, es un deber médico y social el adecuado control de los pacientes con dislipemias. Sin embargo, este control es proporcionalmente escaso y más aún en prevención secundaria, a pesar de que el tratamiento prescripto es generalmente amplio (7). Esto implica una intervención falaz ya sea porque la medicación es inapropiada en tipo y dosis, o porque existe una falta de adherencia y/o seguimiento de los pacientes, verdaderos problemas epidemiológicos y de costos en salud.
Puesto que la forma más común de dislipemia es la hipercolesterolemia, la introducción de las estatinas para su tratamiento a partir de la última década del siglo pasado significó una bisagra en la epidemiología cardiovascular debido a la notable reducción de los eventos cardiovasculares isquémicos (7-10). No obstante, a pesar de las tres décadas pasadas hay pacientes que presentan formas primarias sumamente resistentes al tratamiento con estos fármacos, o que aún con reducción sus lípidos siguen con riesgo residual por razones no bien comprendidas o que, por situaciones particulares, condiciones críticas, disfunción orgánica o interacciones medicamentosas, directamente no pueden recibir estatinas.
Como la investigación farmacológica no cesa y siempre se descubren nuevos mecanismos fisiopatológicos donde intervenir, ante los problemas mencionados en el párrafo anterior han surgido nuevos fármacos (sean alternativos o combinables con los disponibles) para tratar las hiperlipemias como, por ejemplo, los anticuerpos monoclonales, alirocumab y evolocumab; los oligonucleótidos antisentido, inclisirán y volanesorsén y el lípido, ácido bempedoico (6, 7).
En este contexto, el ácido bempedoico resulta una molécula nueva de reciente introducción. Pero a diferencia de las otras no es un compuesto biológico. Por lo tanto, y ante la novedad terapéutica, es objetivo de esta revisión analizar la farmacología experimental y clínica del ácido bempedoico y su papel en la terapia actual y futura de las hiperlipemias, ya sea solo o asociado a otros fármacos.
Fisiopatología de las lipoproteínas y problemas asociados
Los lípidos resultan una de las cuatro biomoléculas fundamentales para los seres vivos (11). Sin ellos, las células como entidad biológica no existirían y la compartimentalización que caracteriza a los eucariotes tampoco, puesto que los agregados fosfolipídicos con colesterol y proteínas generan las biomembranas limitantes. Además, los ácidos grasos son combustibles de alto rendimiento energético cuyo depósito organizado como triglicéridos se desarrolla en el tejido adiposo, tejido especialmente diseñado puesto que es también protector mecánico y térmico. Por último, muchos lípidos son moléculas señal específicas que a través de sus receptores controlan las funciones celulares y el metabolismo en forma local o general, por ejemplo, los ácidos grasos poliinsaturados libres y sus hidroperoxil derivados, los oxiesteroles, las ceramidas, los eicosanoides, los endocannabinoides, la hormonas esteroides y los neuroesteroides.
Los lípidos son insolubles en los medios acuosos predominantes en el organismo, lo que genera un problema porque su distribución entre los compartimientos corporales se hace necesariamente mediante la participación proteínas específicas de unión-transporte y algunas pueden presentar polimorfismos disfuncionales que afecten tal distribución y generen una disposición anormal de los mismos con inadecuado depósito.
En el plasma viajan como lipoproteínas (12) que son micelas fosfolipídicas especiales estabilizadas por apolipoproteínas. Según se ve en la tabla 1, hay cinco tipos o fracciones caracterizadas por su tamaño, densidad y contenido lipídico predominante, los quilomicrones o QM, las lipoproteínas de muy baja densidad o VLDL, las lipoproteínas de densidad intermedia o IDL, las lipoproteínas de baja densidad o LDL y su variante la lipoproteína (a) o Lp (a), y las lipoproteínas de alta densidad o HDL (12, 13). La tabla 2 muestra las diferentes apolipoproteínas (14) o porciones proteicas de las lipoproteínas. Estas participan en la construcción, síntesis, direccionamiento tisular y catabolismo de las distintas fracciones tal como se resume en la figura 1.
Sucintamente, según el lípido predominante hay dos grupos de lipoproteínas, las ricas en triglicéridos, QM, VLDL e IDL, y las ricas en colesterol, LDL y HDL. Pero también se pueden reconocer funcionalmente las lipoproteínas construidas sobre las apo B (como QM, VLDL, IDL y LDL) que transportan triglicéridos y colesterol desde hígado o intestino hacia los demás tejidos, y las construidas sobre apo A-I (como las HDL) que transportan colesterol desde los tejidos hacia el hígado (14).
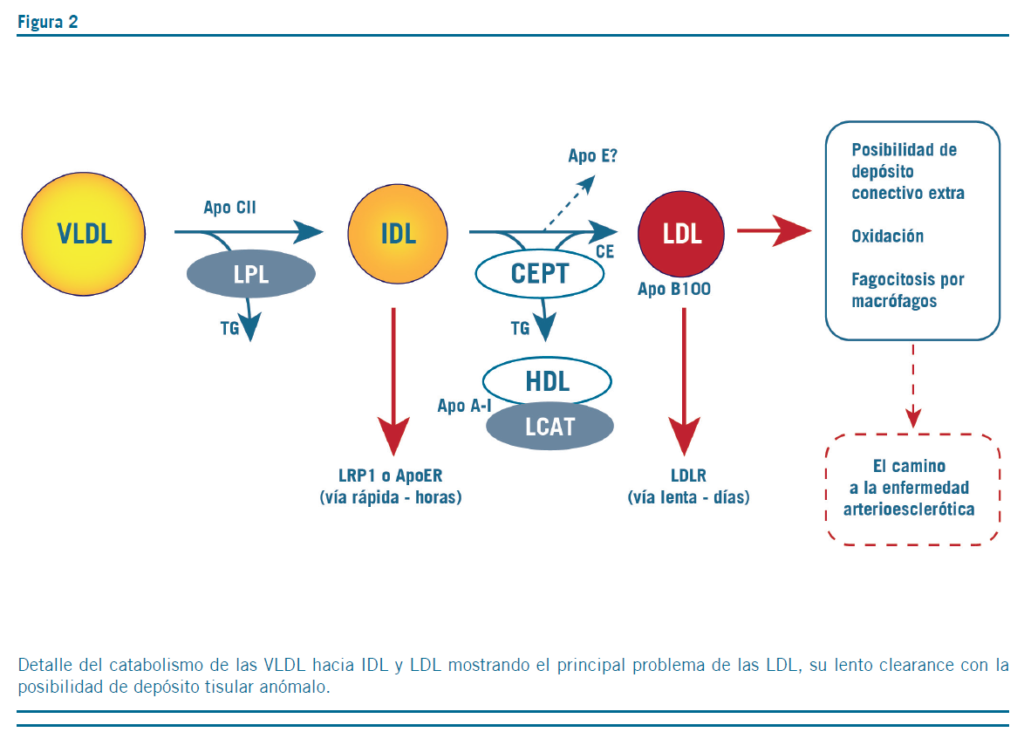
El catabolismo de cada fracción muestra un segundo problema ya que algunas lipoproteínas (como los QM o VLDL) son aclaradas en horas mientras que otras (especialmente las LDL) lo son en días, diferencia que condiciona otra vez la posibilidad de un depósito anormal (ver tabla 1 y figura 2). Por ello, si se incrementa la fracción VLDL es muy probable que aumente también la fracción LDL, que independientemente de su contenido lipídico aumenta el riesgo aterogénico, tal lo observado en la hipertrigliceridemia (7) que condiciona la formación de LDL densas y pequeñas o sdLDL tan aterogénicas como las ricas en colesterol.
Los acúmulos anormales de lipoproteínas ricas en colesterol suelen darse en tejidos conectivos bajo situaciones donde su producción es excesiva o su clearance es sumamente inefectivo, de ahí que no solo haya depósitos vasculares característicos de las placas de ateroma (donde factores hemorreológicos conspiran también para su formación), sino también subcutáneos o articulares en forma de xantelasmas o xantomas respectivamente (15).
Aquí surge un tercer problema, el colesterol una vez sintetizado no es destruible pues el ser humano carece de enzimas para degradar la estructura del ciclopentanoperhidrofenatreno. En consecuencia, la eliminación del colesterol únicamente ocurre por descamación cutánea o mucosa, secreción sebácea y excreción directa bilio-fecal (16). Así, en los depósitos anormales se hallan infiltrados inflamatorios de células espumosas o macrófagos modificados llenos de vesículas cargadas de colesterol que no han podido procesar pues solo lo despachan lentamente vía HDL (ver figura 1C) y que conducen al avance del proceso si la dislipemia causal no se corrige (Figura 2).
Tal progreso puede reseñarse de la siguiente forma, las LDL por su tamaño pasan fácilmente a la íntima sobre todo en zonas de flujo turbulento o donde el endotelio falla en su producción de NO (óxido nítrico) o PGI2 (prostaciclina). Es fácil también que queden atrapadas en la matriz de proteoglicanos mediante interacciones inespecíficas con sus componentes y que las cadenas de ácidos grasos poliinsaturados de los fosfolípidos se oxiden dando productos de peroxidación y malondialdehido sustancias que atraen macrófagos. Inicialmente se forma en la zona una estría grasa, pero con el paso del tiempo, la fagocitosis infructuosa, la inflamación y el estrés oxidativo tal estría deviene en una placa ateromatosa. Placa que posteriormente crecerá por más depósito lipoproteico y fibrosis reactiva pudiendo dar dos resultados evolutivos: una placa estable, consolidada que va obstruyendo lentamente el vaso y genera sintomatología anginosa, o una placa inestable con endotelio friable que al romperse bruscamente genera un episodio isquémico agudo por trombosis o embolia a distancia.

Las bases bioquímicas de la hipertrigliceridemia y de la hipercolesterolemia
El plan corporal que define el hipotálamo presenta ciclos de ingesta-ayuno donde incorporar los nutrientes necesarios para el crecimiento y desarrollo. Como en la naturaleza estos nutrientes no siempre están disponibles o la actividad animal implica migraciones y otras conductas los periodos de ayuno pueden prolongarse por lo que evolutivamente surgieron mecanismos para reservar nutrientes y energía durante periodos de bonanza para ser usados durante la hambruna (fenotipo ahorrador). Estos mecanismos están muy finamente regulados, e incluso son redundantes y cruzados para operar como capas a diferentes niveles (intercelular → hormonas y circuitos neuronales, intracelular → receptores con segundos y terceros mensajeros, y en las propias vías metabólicas → productos finales y moduladores alostéricos) (4).
Como resultado de la industrialización, el ser humano se ha sedentarizado y ha accedido a comidas baratas muy sabrosas, hipercalóricas y poco saciantes. Sin embargo, como no ha perdido el fenotipo ahorrador es sumamente propenso a la obesidad y al síndrome metabólico (4, 5, 17) si no cuida su balance energético.
Los ácidos grasos son cadenas lineales o alifáticas de átomos de carbono que en uno de sus extremos presentan un carboxilo, grupo que les otorga su carácter (11). La cadena lineal varía en tamaño conteniendo casi siempre un número par de átomos ya que su síntesis y elongación se hace en los mamíferos a expensas de acetato que tiene 2 carbonos, y puede ser saturada (es decir el resto de las valencias del carbono son ocupadas con hidrógenos) o insaturada (presenta dobles enlaces cis intercarbono en ciertas posiciones). Si su longitud supera los 4 átomos hará que el ácido graso sea insoluble en agua, pero como su extremo carboxilo está cargado el ácido conserva propiedades anfipáticas y detersivas y tiende a formar micelas.
Los ácidos grasos son una fuente energética importante, incluso rinden más energía por mol que la glucosa, pues están reducidos. Por ello, todo exceso calórico en la ingesta, principalmente carbohidratos, es redirigido a la síntesis de ácidos grasos, fenómeno propiciado por la insulina, mientras que en el ayuno o ante bajas temperaturas se induce su liberación desde el tejido adiposo y el consumo por casi todos los tejidos mediante la b-oxidación mitocondrial, fenómeno estimulado por glucagón y catecolaminas (17, 18). Este balance energético alternativo a la glucosa permite mantener la glucemia para tejidos que solo emplean el azúcar como combustible, p ej., eritrocitos o tejido nervioso.
Valga esta descripción para considerar que los ácidos grasos como tales, es decir libres, son escasos pues en los entornos celulares si no siguen alguna vía de señalización particular (eicosanoides, endocannabinoides) se activan indefectiblemente por unión a la Coenzima A. En forma de acil CoA son rápidamente conducidos hacia su esterificación (formación de enlaces éster entre un ácido y un alcohol) o su consumo.
La esterificación más relevante es con glicerol para formar triglicéridos de depósito o fosfoglicéridos membranares, aunque también se esterifican con colesterol. Esta esterificación ocurre en el retículo endoplásmico y membrana mitocondrial externa de las células y es la que perpetúa la construcción de las membranas biológicas. La síntesis de triglicéridos se llama lipogénesis y tiene lugar en los adipocitos principalmente (puede decirse que esta es una de sus funciones primordiales), en los hepatocitos y en los enterocitos. Los triglicéridos se sintetizan a partir de fosfatidato un precursor común con los fosfoglicéridos y para ello requieren acil CoA (sea de ácidos grasos provenientes de las lipoproteínas o de la síntesis de novo) y glicerol fosfato derivado de la glucólisis (figura 3) (19-21). La única diferencia entre el tejido adiposo y el eje hepatointestinal es que el primero almacena los triglicéridos resultantes mientras que el segundo los coloca en lipoproteínas ricas en triglicéridos para enviarlos a los tejidos.
Entonces, post ingesta y a expensas de la glucosa, hay un acople finamente regulado entre la síntesis de ácidos grasos, la lipogénesis y la formación de lipoproteínas ricas en estos lípidos (especialmente VLDL) como forma de vehiculizar el excedente energético desde el eje hepatointestinal hacia el depósito estable que representa el tejido adiposo o el consumo por varios tejidos especialmente el muscular (18). Si este acople falla en algún punto aparece hipertrigliceridemia o alternativamente esteatosis hepática (4, 5, 17, 18, 21-23):
La hipertrigliceridemia puede surgir tras un exceso sostenido en la ingesta calórica que aumenta la producción hepatointestinal de VLDL. Normalmente, los glúcidos dietarios (glucosa y fructosa) y la insulina inducen las enzimas lipogéneticas y formadoras de mielina a través de los receptores/ intensificadores nucleares ChREBP (proteína de unión al elemento de repuesta a los carbohidratos) y SREBP-1c (proteína de unión al elemento de respuesta a esteroles) respectivamente. De esta forma los glúcidos a nivel hepatointestinal son convertidos en triglicéridos y empaquetados en lipoproteínas. El exceso dietario en glúcidos, lípidos y aminoácidos e incluso alcohol sobreactiva estas señales aún bajo insulino resistencia ya que SREBP-1c puede ser activado por mTORc1 (complejo 1 de la proteína de mamíferos blanco de la rapamicina, un multímero quinasa activadora global del anabolismo que responde entre otras señales a aminoácidos ramificados) lo que determina más síntesis y secreción de VLDL. Una variante ante hipovitaminosis B, falta de colina o una alteración genética es el hígado graso ya que no se forman las micelas fosfolipídicas originarias de las lipoproteínas. Esta situación retiene triglicéridos que tienden a acumularse en forma de gotas grasas y a la vez causa estrés del retículo endoplásmico que retroalimenta el cuadro pues estimula aún más SREBP- 1c vía chaperonas del retículo endoplásmico como p ej., BiP.
La insulino resistencia es la causa principal de hipertrigliceridemia. Esta condición propia del síndrome metabólico y diabetes tipo 2 produce un aumento significativo en la producción de ácidos grasos libres en plasma por parte del tejido adiposo. Puesto que usualmente la insulina inhibe la lipolisis o degradación de triglicéridos estimulada por glucagón o catecolaminas, su déficit la potencia. Este exceso de ácidos grasos es tomado por el hígado y convertido en triglicéridos que, según lo relatado en el párrafo anterior aumentará los niveles de VLDL o la posibilidad de esteatosis. Además, como la insulina resulta fundamental para la secreción-maduración-actividad de la lipoproteín lipasa (LPL, enzima que degrada las lipoproteínas ricas en triglicéridos), en insulino resistencia, cae la actividad de la LPL y se establece una segunda instancia para la hipertrigliceridemia.


Las hipertrigliceridemias familiares severas presentan déficit genético de LPL o de alguna de sus proteínas de control. La LPL es producida por las células de estirpe conectiva (tejido adiposo, conjuntivo, muscular) y secretada al medio extracelular. La proteína LMF1 (factor 1 de maduración de lipasa) acompaña a la LPL durante todo el proceso secretor y permite que alcance su actividad óptima. Una vez secretada se posiciona sobre la cara sanguínea del endotelio vascular gracias a la proteína de anclaje GPIHBP1 en espera de sus sustratos, los triglicéridos contenidos en los QM y VLDL. Finalmente, la actividad hidrolítica requiere de apo C-II y apo A-V presentes en estas lipoproteínas. Entonces cualquier polimorfismo que menoscabe la función de alguna de las proteínas mencionadas anulará el catabolismo de las lipoproteínas ricas en triglicéridos produciendo hipertrigliceridemias muy altas. Si bien, existen inhibidores circulantes de la LPL como apo C-III y las ANGPTL (proteínas símiles a la angiopoyetina) 3, 4 y 8, estos no parecen intervenir en la hipertrigliceridemias familiares, pero si ofrecen oportunidades terapéuticas cuya discusión se halla fuera del objetivo de esta revisión.
Una disbiosis intestinal puede causar hipertrigliceridemia como parte del cuadro adiposo-metabólico. Bajo esta idea, la microbiota disfuncional podría gestionar adiposidad-obesidad por mecanismos energéticos, como un mayor aprovechamiento de ácidos grasos de cadena corta derivados del metabolismo de la fibra celulósica; por anulación del efecto incretina, al alterar la secreción del polipéptido YY (PYY) y de los péptidos símil glucagón 1 y 2 (GLP-1 y GLP-2), o por inhibir la LPL al aumentar expresión del gen de ANGPTL 4.

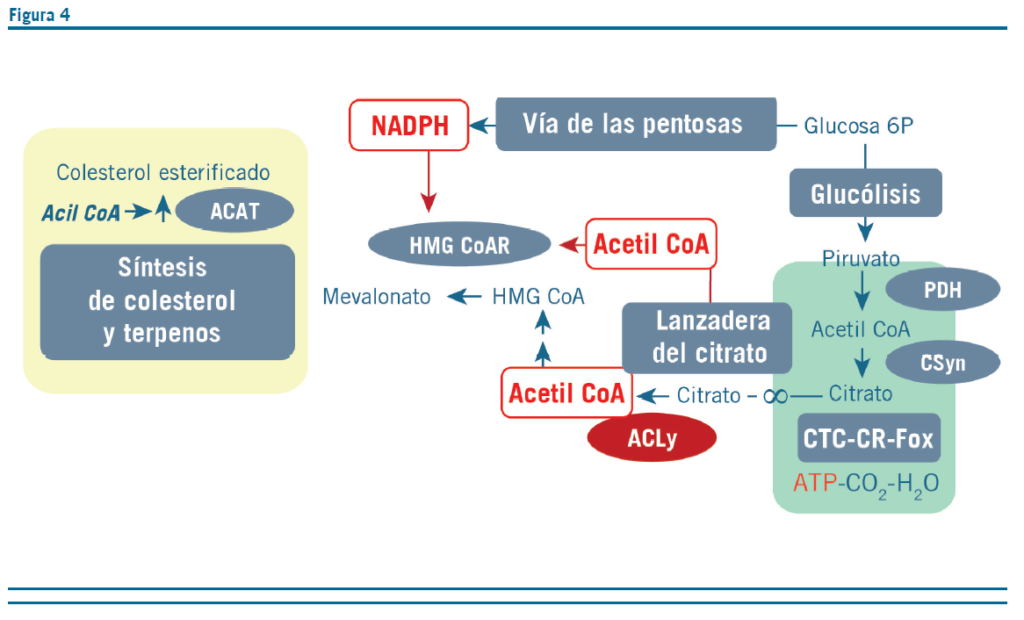
El colesterol, además de su papel como precursor de sales biliares, neuroesteroides, corticoides y hormonas sexuales, resulta un lípido fundamental para la fluidez de las membranas biológicas y el funcionamiento de las proteínas asociadas a ellas. La producción diaria de esta molécula es escasa, cerca de 1 g (16), su síntesis se lleva a cabo en todas las células del organismo siguiendo un ritmo circadiano. El hígado es el órgano con mayor capacidad sintética pues presenta los mayores requerimientos (por las sales biliares) a la vez que resulta el medio excretor de este lípido. Sin embargo, el sistema nervioso, la corteza adrenal y la gónadas son otros órganos con altos requerimientos, por esta razón existe una redistribución intertisular mediado por las lipoproteínas ricas en colesterol (Figura 1). La síntesis de colesterol y de los terpenos intermedios también requiere de acetil CoA citosólica (figura 4) pero a diferencia de la síntesis de ácidos grasos no depende directamente del estímulo insulínico o del consumo de glucosa.
La síntesis de colesterol, y especialmente la hepática, está precisamente regulada, cualquier exceso intracelular del esterol o sus productos inhibe fuertemente la expresión de la enzima clave HMG CoA (hidroximetilglutaril CoA) reductasa y la exposición de los receptores para LDL (LDLR) vía secuestro del intensificador SREBP2 en el retículo endoplásmico (16).
Las hipercolesterolemias pueden deberse a una hiperproducción de las lipoproteínas ricas en colesterol, específicamente la fracción LDL a partir de la fracción VLDL por intercambio plasmático (24) o ser resultado de una falla en la captación y catabolismo de esta fracción más que de un incremento de la síntesis intracelular de colesterol (15). Al respecto, debe entenderse que, si un paciente por razones anteriormente descriptas produce excesivamente VLDL, tiene mayores posibilidades de generar LDL e incluso sdLDL con lo cual junto a hipertrigliceridemia tendrá hipercolesterolemia, pero sobre todo presentará un riesgo de ateromatosis y enfermedad cardiovascular asociada mucho mayor.
Genéticamente, en las hipercolesterolemias familiares, las fallas se centran en los LDLR, en la apo B que lo reconoce y en la endoproteasa que inactiva los LDLR, la PCSK9 (proprotein convertase subtilisin/kexin 9). Justamente, el tratamiento con estatinas aumenta notablemente la expresión de los LDLR en el hígado, pero como en las formas familiares el receptor no funciona, este tratamiento responde muy poco o directamente fracasa.
Finalmente, debe considerarse que si un paciente con hipercolesterolemia recibe una dieta rica en colesterol es probable el agravamiento del cuadro, aun cuando el aporte de colesterol dietario solo implica un 10% del total diario, debido a la redistribución en las fracciones VLDL-LDL.
Bibliografía sugerida
• 1. Iummato LE, Blanco J, Goldberg L, King A, Rodríguez Cámara MJ. Tendencias de la mortalidad por enfermedades no transmisibles en Argentina entre 1997 y 2021. Rev Argent Salud Publica 2024; 16: e123.
• 2. Diaz A, Espeche W, March C, Flores R, Parodi R, Genesio MA y col. Prevalencia del síndrome metabólico en Argentina en los últimos 25 años: Revisión sistemática de estudios observacionales poblacionales. Hipertens Riesgo Vasc 2018; 35: 64-9. doi: 10.1016/j.hipert.2017.08.003.
• 3. McLaughlin T, Abbasi F, Cheal K, Chu J, Lamendola C, Reaven G. Use of metabolic markers to identify overweight individuals who are insulin resistant. Ann Intern Med 2003; 139: 802-9. doi: 10.7326/0003-4819-139-10-200311180-00007.
• 4. Serra HA, Fadel DO. Síndrome metabólico y psicofármacos, un fenómeno creciente y peligroso. Parte 1. Rev Soc Arg Endocrinol Ginecol Reproductiva 2017; 24: 26-36.
• 5. Serra HA, Fadel DO. Síndrome metabólico y psicofármacos, un fenómeno creciente y peligroso. Parte 2. Rev Soc Arg Endocrinol Ginecol Reproductiva 2017; 24: 66-72.
• 6. Elikir G, Cúneo C, Lorenzatti A, Aimone D, Berg G, Corral P y col, eds. Guía de Práctica Clínica de la Sociedad Argentina de Lípidos sobre Diagnóstico y Tratamiento de las Dislipemias en Adultos 2019. Córdoba: Sociedad Argentina de Lípidos 2019.
• 7. de Abajo Olea S. Epidemiología, definición, clasificación, despistaje y diagnóstico de las dislipemias. SEMERGEN 2009; 35 Supl 3: 3-9.
• 8. Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur Heart J 2020; 41: 111-88. doi: 10.1093/eurheartj/ehz455. Erratum in: Eur Heart J 2020; 41: 4255. doi: 10.1093/eurheartj/ehz826.
• 9. Grundy SM, Cleeman JI, Merz CN, Brewer HB Jr, Clark LT, Hunninghake DB, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110: 227-39. doi: 10.1161/01.CIR.0000133317.49796.0E. Erratum in: Circulation 2004; 110: 763.
• 10. Clarke R, Von Ende A, Schmidt LE, Yin X, Hill M, Hughes AD, et al. Apolipoprotein proteomics for residual lipid related risk in coronary heart disease. Circ Res 2023; 132: 452-64. doi: 10.1161/CIRCRESAHA.122.321690.
• 11. Botham KM, Mayes PA. Chapter 21 Lipids of Physiological Importance. In Kennelly PJ, Botham KM, McGuinness OP, Rodwell VW, Weil PA, eds, Harper’s Illustrated Biochemistry 32nd ed. New York: Lange Mc Graw Hill 2023; pp 205-16.
• 12. Lent-Schochet D, Jialal I. Biochemistry, Lipoprotein Metabolism. [Updated 2023 Jan 16]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK553193/.
• 13. Corral P, Nogueira JP, Schreier L, Berg G, Masson W, Aguilar Salinas C y col. Documento de posición de la Sociedad Argentina de Lípidos (SAL) sobre la lipoproteína (a) o Lp(a). Rev Arg de Lípidos 2022; 6: 39-49.
• 14. Mehta A, Shapiro MD. Apolipoproteins in vascular biology and atherosclerotic disease. Nat Rev Cardiol 2022; 19: 168-79. doi: 10.1038/s41569- 021-00613-5.
• 15. Araujo M, Arrupe M, Bañares V, Berg G, Corral P, Cuartas S y col. Documento de posición. Manejo del paciente con hipercolesterolemia familiar homocigota. Rev Arg de Lípidos 2024; 8: 62-74.
• 16. Botham KM, Mayes PA. Chapter 26 Cholesterol Synthesis, Transport, & Excretion. In Kennelly PJ, Botham KM, McGuinness OP, Rodwell VW, Weil PA, eds, Harper’s Illustrated Biochemistry 32nd ed. New York: Lange Mc Graw Hill 2023; pp 259-72.
• 17. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev 2018; 98: 2133-223. doi:10.1152/physrev.00063.2017.
• 18. Alves-Bezerra M, Cohen DE. Triglyceride Metabolism in the Liver. Compr Physiol 2017; 8: 1-8. doi: 10.1002/cphy.c170012.
• 19. Botham KM, Mayes PA. Chapter 23 Biosynthesis of Fatty Acids & Eicosanoids. In Kennelly PJ, Botham KM, McGuinness OP, Rodwell VW, Weil PA, eds, Harper’s Illustrated Biochemistry 32nd ed. New York: Lange Mc Graw Hill 2023; pp 226-38.
• 20. Botham KM, Mayes PA. Chapter 24 Metabolism of Acylglycerols & Sphingolipids. In Kennelly PJ, Botham KM, McGuinness OP, Rodwell VW, Weil PA, eds, Harper’s Illustrated Biochemistry 32nd ed. New York: Lange Mc Graw Hill 2023; pp 239-46.
• 21. Corral P, Masson W, Cafferata A, Closs C, Bañares V, Schreier L y col. Diagnóstico y tratamiento de la hipertrigliceridemia grave: Documento de posición. Rev Arg de Lípidos 2021; 5: 25-36.
• 22. Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: An integrative view. Cell 2012; 148:1258-70. doi: 10.1016/j.cell.2012.01.035.
• 23. Greiner T, Bäckhed F. Effects of the gut microbiota on obesity and glucose homeostasis. Trends Endocrinol Metab 2011; 22:117-23. doi: 10.1016/j.tem.2011.01.002.
• 24. Nelson AJ, Sniderman AD, Ditmarsch M, Dicklin MR, Nicholls SJ, Davidson MH, Kastelein JJP. Cholesteryl ester transfer protein inhibition reduces major adverse cardiovascular events by lowering apolipoprotein B levels. Int J Mol Sci 2022; 23: 9417. doi: 10.3390/ijms23169417.





No comments! Be the first commenter?